Manipulating Large Molecule Files¶
Problem¶
You want to manipulate large molecule files.
Ingredients¶
|

Solution¶
This recipe discusses several examples that handle large molecule files:
- Creating Molecule Database Index File
- Counting Molecules
- Output Molecule Titles
- Extracting Molecules by Title
- Extracting Random Set of Molecules
- Splitting Molecule Database
- Sorting Molecules
All solutions presented here utilize the OEMolDatabase class that provides fast read-only random access to molecular file formats that are readable by OEChem TK.
Hint
The usage of OEMolDatabase is highly recommended when:
- You want to manipulate molecule files without parsing the molecules.
- You want to access molecules in a file randomly.
- You want to handle a large set of molecules that can not be held in memory all at once.
Creating Molecule Database Index File¶
The first simple example shows how to create an index file by calling the OECreateMolDatabaseIdx function. The OEGetMolDatabaseIdxFileName function returns the name of the index file associated with the given molecule filename.
The index file of a molecule database stores file position offsets of the molecules in the file. Generating an index file can be expensive, but it can be created only once and then it can speed up the handling of large molecule files significantly.
1 2 3 4 5 6 7 | def CreateMolDatabaseIndexFile(ifname):
idxfname = oechem.OEGetMolDatabaseIdxFileName(ifname)
if os.path.exists(idxfname):
oechem.OEThrow.Warning("%s index file already exists" % idxfname)
elif not oechem. OECreateMolDatabaseIdx(ifname):
oechem.OEThrow.Warning("Unable to create %s molecule index file", idxfname)
|
Download code
moldb_create.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_create.py drugs.sdf
Running the above command will generate the drugs.sdf.idx molecule database index file.
See also
- Index files section in the OEChem TK manual
Counting Molecules¶
The first is a simple example that show how to count the number of molecules in a file. After initializing an OEMolDatabase object with an input stream, the OEMolDatabase.NumMols method returns the number of molecule records read from the file into the database.
1 2 3 4 5 6 7 8 9 10 11 | def MolCount(fname):
ifs = oechem.oemolistream()
if not ifs.open(fname):
oechem.OEThrow.Warning("Unable to open %s for reading" % fname)
return 0
moldb = oechem.OEMolDatabase(ifs)
nummols = moldb.NumMols()
print("%s contains %d molecule(s)." % (fname, nummols))
return nummols
|
Download code
moldb_molcount.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_molcount.py drugs.sdf
Running the above command will generate the following output:
drugs.sdf contains 6 molecule(s).
===========================================================
Total 6 molecules
When the drugs.sdf file is opened by the molecule database, it automatically detects the presence of the corresponding index file drugs.sdf.idx. In case when the index file exists, the number of file offsets stored in the index files equal to the number of molecules stored in the corresponding drugs.sdf molecule file. So returning the number of molecules stored in the drugs.sdf file does not require to actually reading and parsing the molecules stored inside.
Output Molecule Titles¶
The next example outputs the title of the molecules stored in a file. After initializing an OEMolDatabase object, the molecules stored in the database can be accessed by their index. The title of all molecules can be retrieved by looping over the indices and calling the OEMolDatabase.GetTitle method that returns the title of molecule at the given index.
1 2 3 4 5 6 7 8 | def OutputMolTitles(ifs, ofs):
moldb = oechem.OEMolDatabase(ifs)
for idx in range(moldb.GetMaxMolIdx()):
title = moldb.GetTitle(idx)
if len(title) == 0:
title = "untitled"
ofs.write('%s\n' % title)
|
Download code
moldb_gettitles.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_gettitles.py drugs.sdf
Running the above command will generate the following output:
acetsali
acyclovi
alprenol
aminopy
atenolol
caffeine
Extracting Molecules by Title¶
The next example extracts compound(s) from a file based on a molecule title or a set of titles. This example uses the same OEMolDatabase.GetTitle method to access the title of the molecules. Molecules with matching title are then written directly to the output stream by using the OEMolDatabase.WriteMolecule method.
This example illustrates one of the main advantages of using molecule databases. Extracting molecules this way does not require actually parsing the molecules. The code below simply copies the bytes that encode the molecules from an input stream to an output stream without ever calling OEReadMolecule or OEWriteMolecule.
1 2 3 4 5 6 7 | def MolExtract(ifs, ofs, titleset):
moldb = oechem.OEMolDatabase(ifs)
for idx in range(moldb.GetMaxMolIdx()):
title = moldb.GetTitle(idx)
if title in titleset:
moldb.WriteMolecule(ofs, idx)
|
Download code
moldb_molextract.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_molextract.py -in drugs.sdf -title aminopy -out .sdf | python3 moldb_gettitles.py .sdf
Running the above command will generate the following output:
aminopy
Extracting Random Set of Molecules¶
It is a frequently occurring task to extract a random set of molecules from a large molecule file. The code below first creates a random index set using the random.sample() function. The molecules corresponding to the indices are then copied over from the input stream to the output stream. The random access feature provided by the OEMolDatabase class makes this process very fast. Implementing the same process without OEMolDatabase class would either require holding all molecules in memory at once or in the case of a very large molecule file it would require reading the molecule file more than once.
1 2 3 4 5 6 7 8 9 | def RandomizePercent(ifs, ofs, percent, rand):
moldb = oechem.OEMolDatabase(ifs)
indices = range(moldb.GetMaxMolIdx())
size = max(1, int(percent * 0.01 * moldb.GetMaxMolIdx()))
mlist = rand.sample(indices, size)
WriteDatabase(moldb, ofs, mlist, size)
|
1 2 3 | def WriteDatabase(moldb, ofs, mlist, size):
for molidx in mlist[:size]:
moldb.WriteMolecule(ofs, molidx)
|
Download code
moldb_randomsample.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_randomsample.py -in drugs.sdf -out .sdf -p 50 -seed 1 | python3 moldb_gettitles.py .sdf
Running the above command will generate the following output:
aacetsali
atenolol
aminopy
prompt > python3 moldb_randomsample.py -in drugs.sdf -out .sdf -n 4 -seed 5 | python3 moldb_gettitles.py .sdf
Running the above command will generate the following output:
aminopy
caffeine
atenolol
alprenol
Splitting Molecule Database¶
The next example shows how to split a large molecule file into smaller pieces. There are two possible ways to do this.
The first function SplitChunk shows how to split a molecule into \(n\) sized chunks. Each output file is going to contain \(n\) number of molecules except the last one that will have the remainder.
1 2 3 4 5 6 7 8 9 10 | def SplitChunk(ifs, chunksize, outbase, ext):
moldb = oechem.OEMolDatabase(ifs)
chunk, count = 1, chunksize
for idx in range(moldb.GetMaxMolIdx()):
if count == chunksize:
ofs = NewOutputStream(outbase, ext, chunk)
chunk, count = chunk + 1, 0
count += 1
moldb.WriteMolecule(ofs, idx)
|
The second function SplitNParts shows how to spit a molecule file by determining the number of chunks rather than the size of the chunks.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | def SplitNParts(ifs, nparts, outbase, ext):
moldb = oechem.OEMolDatabase(ifs)
molcount = moldb.NumMols()
chunksize, lft = divmod(molcount, nparts)
if lft != 0:
chunksize += 1
chunk, count = 1, 0
ofs = NewOutputStream(outbase, ext, chunk)
for idx in range(moldb.GetMaxMolIdx()):
count += 1
if count > chunksize:
if chunk == lft:
chunksize -= 1
ofs.close()
chunk, count = chunk + 1, 1
ofs = NewOutputStream(outbase, ext, chunk)
moldb.WriteMolecule(ofs, idx)
|
Download code
moldb_molchunk.py and drugs.sdf supporting data file
Usage:
Splitting a molecule file into chunks of size \(n\)
prompt > python3 moldb_molchunk.py -in drugs.sdf -out chunk.sdf -size 4 | python3 moldb_molcount.py chunk*.sdf
Running the above command will generate the following output:
chunk_0000001.sdf contains 4 molecule(s).
chunk_0000002.sdf contains 2 molecule(s).
===========================================================
Total 6 molecules
Splitting a molecule file into \(n\) chunks:
prompt > python3 moldb_molchunk.py -in drugs.sdf -out chunk.sdf -num 4 | python3 moldb_molcount.py chunk*.sdf
Running the above command will generate the following output:
chunk_0000001.sdf contains 2 molecule(s).
chunk_0000002.sdf contains 2 molecule(s).
chunk_0000003.sdf contains 1 molecule(s).
chunk_0000004.sdf contains 1 molecule(s).
===========================================================
Total 6 molecules
Sorting Molecules¶
The last two examples show how to sort large molecule files. In the first example the sorting criterion is the molecule title of the input compounds. In the SortByTitle function, first a OEMolDatabase object is constructed with an input molecule stream. Then a list of (title - molecule index) tuples is constructed using the OEMolDatabase.GetTitles method that returns all the titles for the database. After sorting this list in alphabetical order, the indices are extracted and the molecules in the database are re-ordered by calling the OEMolDatabase.Order method. Subsequent calls to OEMolDatabase.Save method output the database in this new order.
1 2 3 4 5 6 7 8 9 10 | def SortByTitle(ifs, ofs):
moldb = oechem.OEMolDatabase(ifs)
titles = [(t, i) for i, t in enumerate(moldb.GetTitles())]
titles.sort()
indices = [i for t, i in titles]
moldb.Order(indices)
moldb.Save(ofs)
|
Download code
moldb_titlesort.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_titlesort.py drugs.sdf .sdf | python3 moldb_gettitles.py .sdf
Running the above command will generate the following output:
acetsali
acyclovi
alprenol
aminopy
atenolol
caffeine
The last example shows how to sort molecules by their molecular complexity. In the SortByComplexity function, first a OEMolDatabase object is constructed with an input molecule stream. Then by looping over all entries of the database, a list is generated that stores (complexity score - molecule index) tuples. After sorting this list (by the complexity scores), the indices are extracted and the molecules in the database are re-ordered by calling the OEMolDatabase.Order method. Subsequent calls to OEMolDatabase.Save method output the database in this new order.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | def SortByComplexity(ifs, ofs):
moldb = oechem.OEMolDatabase(ifs)
complexitylist = []
mol = oechem.OEGraphMol()
for idx in range(moldb.GetMaxMolIdx()):
complexscore = float("inf")
if moldb.GetMolecule(mol, idx):
oechem.OEPerceiveChiral(mol)
complexscore = oemedchem.OETotalMolecularComplexity(mol)
complexitylist.append((complexscore, idx))
complexitylist.sort()
indices = [idx for s, idx in complexitylist]
moldb.Order(indices)
moldb.Save(ofs)
|
Download code
moldb_complexitysort.py and drugs.sdf supporting data file
Usage:
prompt > python3 moldb_complexitysort.py drugs.sdf .sdf | python3 moldb_gettitles.py .sdf
Running the above command will generate the following output:
acetsali
caffeine
acyclovi
aminopy
alprenol
atenolol
Hint
The OEMolDatabase.Save method is optimized for the case whenever the output file format is the same as the input file format used to initialize the database. If the same file format is used, the molecule record’s bytes will be streamed directly, without an intermediate OEMolBase to do the conversion.
Discussion¶
The above examples perform various tasks of manipulating molecule files. Most of the scripts described here do no actually require fully parsing the molecules. The OEMolDatabase class was implemented for the purpose of performing these kind of operations very rapidly. Figure 1 illustrates the performance improvement that can be achieved when using molecule databases.
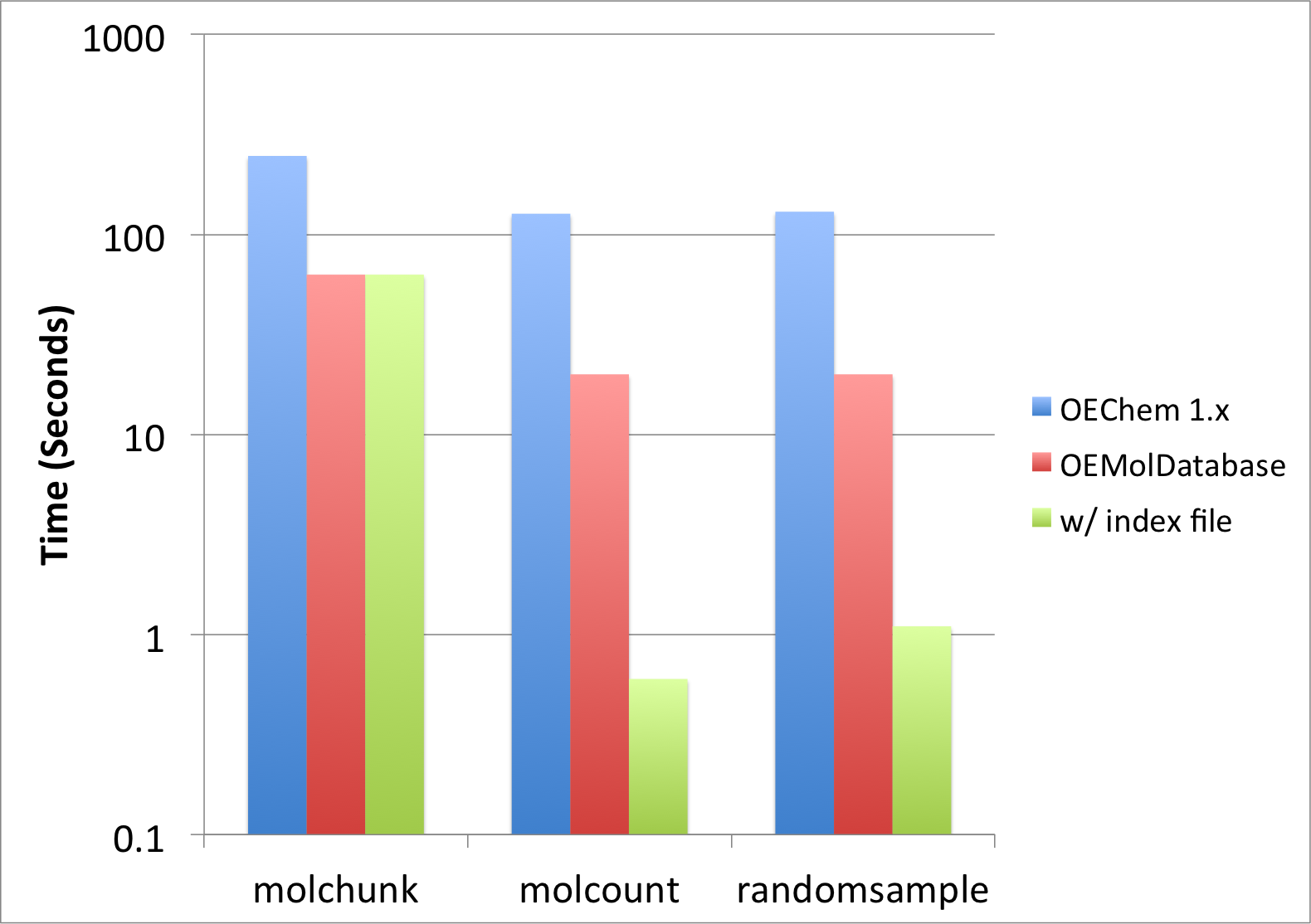
Figure 1: Performance improvement when using OEMolDatabase
The molecule file drugs.sdf (containing only 6 molecules) used in this recipe to show the usage of the examples does not illustrate how awesome and powerful molecule databases are.
See also
- Rapid Similarity Searching of Large Molecule Files and Similarity Searching of Large Molecule Files recipes that show how to perform 2D similarity search on large molecule files.
See also in Python documentation¶
See also in OEChem TK manual¶
Theory
- Molecular Database Handling chapter
API
- OECreateMolDatabaseIdx function
- OEGetMolDatabaseIdxFileName function
- OEMolDatabase class