OEToolkits 2014.Oct¶
This release of the OpenEye toolkits contains many new features and bug fixes. The most important have been highlighted here, the full list can be accessed below.
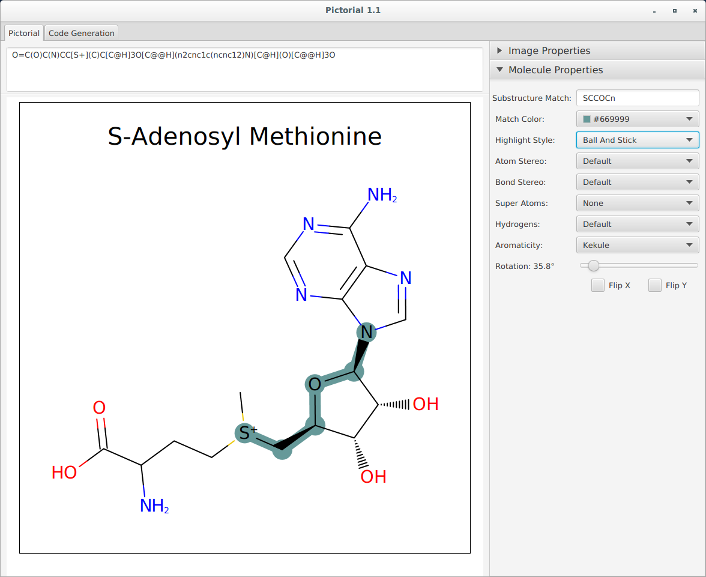
Pictorial application for automatically writing OEDepict code¶
OEDepict TK is a powerful API for the “2D” depiction of small molecules. While such depictions are universal in chemistry there are many variants in drawing style, the majority of which are captured in OEDepict’s extensive set of options. To aid navigation through the many options available we now provide a small application, Pictorial, written in Java, that will autogenerate code that can then be used in your own programming. The figure below is an illustration of the interface.
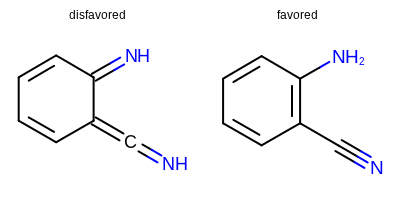
Major improvements to Quacpac’s protomer assignment algorithms¶
Quacpac TK is a set of tools for the assignment of charges, ionization states and proton positions. The latter has been significantly improved to produce more reasonable tautomeric forms, for instance by disfavoring exocyclic bonds adjacent to aromatic bonds (e.g. favoring aromatic forms). An example is depicted below.
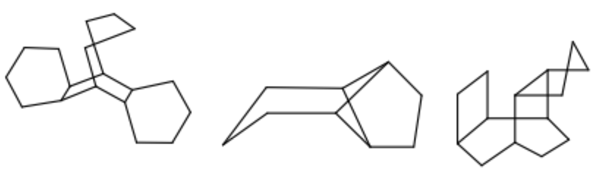
Major improvements to depiction of complex ring systems¶
Depiction of complex molecules in 2D is both a science and an art. Algorithms help with the majority of cases but sometimes they have to be supplemented with the ‘art’ of hand-drawn templates. In our continuing quest to provide the best 2D depiction of molecules, this release includes an additional 550 such templates to OEDepict and 2D coordinate generation in OEChem. Some examples are shown below.
Multi-precision conformer handling¶
There are times where precision is important and times when, due to memory consumption, it gets in the way of computation. As such, this release of the toolkits includes two new levels of precision. In OEChem the basic molecular object OEMol can now store conformer coordinates as 16-bit ‘half-floats’, 64-bit double precision number or long doubles (typically 80 bits although storage requirements may vary). Half-floats have five bits reserved for the exponent, eleven for the fraction. Examples of where it might be useful include shape representation (robust to small coordinate changes) or compact storage. Extra-precision is useful when interfacing with quantum programs or for storage in rotor-offset form in OEB files.
Warning
C++ users may notice that OEChem::OEMCMolBaseT and OEChem::OEConfBaseT have been renamed to OEMCMolBase and OEConfBase respectively. This change was to enable the above feature and to ease the use of the documentation by Python, Java, and C# users who should see no effect from this change.
New force field in Szybki¶
Szybki TK encompasses our capabilities for molecular optimization. This version of the toolkit includes our latest advances in the calculation of intermolecular interactions, i.e. the combination of dispersion (attractive), Fermi overlap (repulsion) and Coulombic interactions (either). These are captured in the IEFF formulation, which represents the first two interaction terms in more detail than typical VdW forms. In addition, Coulombic interactions are calculated using atom-centered point multipoles, as output by quantum chemistry codes. OpenEye provides facilities to input these into OEB files for proteins and ligands, allowing a new level of sophistication in the evaluation of complex interactions. In addition, the Szybki toolkit also includes a new function call to allow for specifically constraining a single torsion and new methods to assign flexibility to residues during protein-ligand optimization.
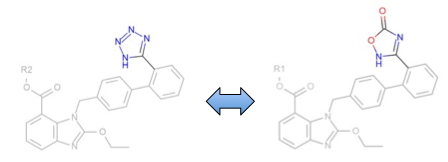
Matched Pairs analysis beta API added to OEMedChem TK¶
This release includes an exciting beta of OEMedChem which includes containing facilities for “Matched Pair” analysis. The matched pair concept has resonated in the medicinal chemistry community because it provides a simple method of comparing two molecules, i.e. ascertaining whether they differ in chemically obvious ways, e.g. a swap of a fluorine for a chlorine or a benzamide for a benzamidine, and then using historical data on such transformations to make predictions on property differences. The functions within the OEMedChem toolkit in this release are capable of searching datasets for such pairings, allowing for such prior expectations to be tabulated. We stress this is a beta release feature and look forward to incorporating community suggestions and requests.
Major bug fixes¶
Warning
It is strongly recommended for users of the 2014.Feb and 2014.Jun releases to upgrade to the 2014.Oct release because of the following major bug fixes for the SDF, MDL, and CDX file formats.
The addition of 2D coordinate generation for the SDF, MDL, and CDX file formats in the 2014.Feb release, OEChem 2.0.0, created the possibility of erroneously changing stereo chemistry in the following cases:
Bond stereo chemistry in macrocycles
Atom stereo from wedges or hashes between atoms with an angle less than 180 degrees
The 2D coordinate generation can not always generate coordinates that represent the required stereo chemistry. For these cases, OEChem will now leave the coordinates in the resulting file all zero. This will make file readers choose to honor the stereo marks instead of the 2D coordinates.
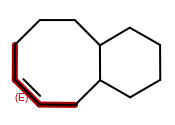
|
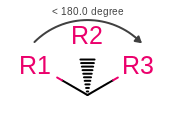
|
[1] |
[2] |
General notices¶
The following is no longer supported on RedHat Enterprise Linux 5:
Python 2.6
gcc 4.3
gcc 4.4
Ubuntu 10.04 is no longer supported.
This will be the last release to support SuSe 11.
OSX 10.7 is no longer supported.
This will be the last release to support Visual Studio 2008 for C++ and C# toolkits.