Pattern Matching
OEChem TK includes facilities to perform different types of pattern (graph) matching. Graph matching is based on node (atom) and edge (bond) correspondences. An atom which satisfies the conditions of a node in a query graph is said to match. Likewise, a bond which satisfies the conditions of an edge in a query graph is said to match. Pattern matching is the process of identifying groupings of matching nodes and edges. Substructure search, or subgraph isomorphism, is the process of finding a graph match which is less than or equal to a larger graph. Maximum common substructure search is the process of identifying the maximal graph correspondence between two graphs. Clique detection is the process of finding all possible correspondences between two graphs within a set of bounds.
Substructure Search
Substructure searches can be done in OEChem TK using the OESubSearch class. The OESubSearch class can be initialized with a SMARTS pattern, an OEQMolBase query molecule, or an OEMolBase with expression options. The following example demonstrates how to initialize a OESubSearch instance with a SMARTS pattern, and perform a substructure search.
Listing 1: Simple substructure search example
from openeye import oechem
mol = oechem.OEGraphMol()
oechem.OESmilesToMol(mol, "c1ccccc1C")
# create a substructure search object
ss = oechem.OESubSearch("c1ccccc1")
oechem.OEPrepareSearch(mol, ss)
if ss.SingleMatch(mol):
print("benzene matches toluene")
else:
print("benzene does not match toluene")
In Listing 1, the query pattern is
benzene and the molecule in which the substructure is being searched
for is toluene. Since benzene is a substructure of toluene the
OESubSearch.SingleMatch method will return
true. The SingleMatch
method returns true if a single subgraph isomorphism is detected
in the molecule passed as the function argument.
Using the SingleMatch
function in Listing 2, it is possible to loop through any molecular
file and extract molecules that match a particular SMARTS
pattern.
Listing 2: Extracting molecules based upon a substructure
from openeye import oechem
import sys
ss = oechem.OESubSearch(sys.argv[1])
ifs = oechem.oemolistream(sys.argv[2])
ofs = oechem.oemolostream(sys.argv[3])
for mol in ifs.GetOEGraphMols():
oechem.OEPrepareSearch(mol, ss)
if ss.SingleMatch(mol):
oechem.OEWriteMolecule(ofs, mol)
The OESubSearch class is able to
identify the atom and bond correspondences of the pattern and target
structures. Listing 3 extends the simple match
example to write out all atom correspondences between benzene and
toluene.
Listing 3: Substructure search with atom mappings
from openeye import oechem
mol = oechem.OEGraphMol()
oechem.OESmilesToMol(mol, "c1ccccc1C")
# create a substructure search object
ss = oechem.OESubSearch("c1ccccc1")
oechem.OEPrepareSearch(mol, ss)
# loop over matches
for count, match in enumerate(ss.Match(mol)):
print("Match", count + 1, ":", end=" ")
print("pattern atoms:", end=" ")
for ma in match.GetAtoms():
print(ma.pattern.GetIdx(), end=" ")
print("target atoms:", end=" ")
for ma in match.GetAtoms():
print(ma.target.GetIdx(), end=" ")
print()
The output of Listing 3 is the following:
Match 1 : pattern atoms: 0 1 2 3 4 5 target atoms: 0 1 2 3 4 5
Match 2 : pattern atoms: 0 1 2 3 4 5 target atoms: 0 5 4 3 2 1
Match 3 : pattern atoms: 0 1 2 3 4 5 target atoms: 1 2 3 4 5 0
Match 4 : pattern atoms: 0 1 2 3 4 5 target atoms: 1 0 5 4 3 2
Match 5 : pattern atoms: 0 1 2 3 4 5 target atoms: 2 3 4 5 0 1
Match 6 : pattern atoms: 0 1 2 3 4 5 target atoms: 2 1 0 5 4 3
Match 7 : pattern atoms: 0 1 2 3 4 5 target atoms: 3 4 5 0 1 2
Match 8 : pattern atoms: 0 1 2 3 4 5 target atoms: 3 2 1 0 5 4
Match 9 : pattern atoms: 0 1 2 3 4 5 target atoms: 4 5 0 1 2 3
Match 10 : pattern atoms: 0 1 2 3 4 5 target atoms: 4 3 2 1 0 5
Match 11 : pattern atoms: 0 1 2 3 4 5 target atoms: 5 0 1 2 3 4
Match 12 : pattern atoms: 0 1 2 3 4 5 target atoms: 5 4 3 2 1 0
The OESubSearch.Match method performs subgraph
isomorphism determination for instances of
OEMolBase or
OEQMolBase and returns an iterator over
all detected subgraphs. Each of the detected subgraphs can be queried
for their atom and bond correspondences. In this particular example,
the benzene substructure is identified twelve times in toluene. There
are twelve matches because the benzene ring can be rotated around for
6 matches, and then flipped and rotated around for another 6 matches,
yielding a total of 12 matches. Each of the matches differ in their
atom and bond correspondences to the pattern substructure.
A match or subgraph is considered unique if it differs from all other
subgraphs found previously by at least one atom. When doing unique
matching, two subgraph matches which cover the same atoms, albeit in
different orders, will be called duplicates and it will be discarded.
In order to retrieve only unique matches, the
Match method has to be called
with a second argument being set to true. In Listing
3, using unique search would yield only a single match for
benzene in toluene.
An OESubSearch may be initialized using a
SMARTS or a query molecule
OEQMolBase. Query molecules must have
atom and bond expressions built for the entire molecule to be able to
initialize the search object (see
OEQMolBase.BuildExpressions in the API).
OESubSearch.GetPattern returns a read-only
reference to the query molecule contained in an instance of
OESubSearch. OEQMolBase methods can
be used to interrogate the returned OEQMolBase
reference.
The OESubSearch.SetMaxMatches method sets the
maximum number of subgraphs to be returned by the
OESubSearch.Match method. Once the maximum number
of subgraphs has been found the search is terminated. By default, an
OESubSearch is constructed with the maximum number of
matches set to 1024. The constraint on the maximum number of matches
can be removed by calling OESubSearch.SetMaxMatches
with a value of 0.
When an OESubSearch object is initialized with a query molecule,
the OESubSearch.Match method returns a match between the copy
of the query molecule stored inside the OESubSearch object and the
target molecule.
The Listing 4 example below
illustrates how to reconstruct matches between the original query
molecule that is used to initialize a search and a target molecule.
In order to keep track of the atoms, each atom of the query molecule is
marked with its own unique atom index before the
OESubSearch object is constructed.
After the matches are returned by calling the
Match the original atoms can be
retrieved by these indices.
Listing 4: Substructures match for query molecule
itag = oechem.OEGetTag("__orig_idx")
for ai in qmol.GetAtoms():
ai.SetData(itag, ai.GetIdx())
ss = oechem.OESubSearch(qmol, oechem.OEExprOpts_DefaultAtoms, oechem.OEExprOpts_DefaultBonds)
tmol = oechem.OEGraphMol()
oechem.OESmilesToMol(tmol, "Cc1ccccc1")
oechem.OEPrepareSearch(tmol, ss)
for mi in ss.Match(tmol, True):
match = oechem.OEMatch()
for apairi in mi.GetAtoms():
pidx = apairi.pattern.GetData(itag)
pattern = qmol.GetAtom(oechem.OEHasAtomIdx(pidx))
match.AddPair(pattern, apairi.target)
See also
Maximum Common Substructure Search
The maximum common substructure (henceforth MCS) of two molecular
graphs can be identified using the OEMCSSearch class.
Listing 5 demonstrates how to initialize an
OEMCSSearch object, perform a maximum common
substructure search, and then retrieve the matches.
Listing 5: Maximum common substructure search
from openeye import oechem
pattern = oechem.OEGraphMol()
target = oechem.OEGraphMol()
oechem.OESmilesToMol(pattern, "c1cc(O)c(O)cc1CCN")
oechem.OESmilesToMol(target, "c1c(O)c(O)c(Cl)cc1CCCBr")
atomexpr = oechem.OEExprOpts_DefaultAtoms
bondexpr = oechem.OEExprOpts_DefaultBonds
# create maximum common substructure object
mcss = oechem.OEMCSSearch(pattern, atomexpr, bondexpr, oechem.OEMCSType_Exhaustive)
# set scoring function
mcss.SetMCSFunc(oechem.OEMCSMaxAtoms())
# ignore matches smaller than 6 atoms
mcss.SetMinAtoms(6)
unique = True
# loop over matches
for count, match in enumerate(mcss.Match(target, unique)):
print("Match %d:" % (count + 1))
print("pattern atoms:", end=" ")
for ma in match.GetAtoms():
print(ma.pattern.GetIdx(), end=" ")
print("\ntarget atoms: ", end=" ")
for ma in match.GetAtoms():
print(ma.target.GetIdx(), end=" ")
# create match subgraph
m = oechem.OEGraphMol()
oechem.OESubsetMol(m, match, True)
print("\nmatch smiles =", oechem.OEMolToSmiles(m))
The first molecule, pattern, is dopamine, and the second molecule,
target, is a dopamine analog. The OEMCSSearch
instance is initialized with the dopamine query, atom and bond
expressions, and the type of the search.
mcss = oechem.OEMCSSearch(pattern, atomexpr, bondexpr, oechem.OEMCSType_Exhaustive)
The atom and bond expressions define criteria for atom and bond
equivalence used during the search and are defined in the
OEExprOpts namespace. See the
OEExprOpts Namespace section for more information.
An OEMCSSearch object can also be constructed from a SMARTS string directly. In this case standard SMARTS matching rules apply for what constitutes a match. If it is constructed with an OEQMolBase, then whatever atom and bond expressions have been applied to the OEQMolBase will apply in the MCS search.
The last argument of the initialization defines the search type,
either Approximate or
Exhaustive. The difference
between the two search types is detailed in the
Exhaustive and approximate MCSS section. This argument is
optional, if it is not specified, then the
exhaustive method is
employed.
During the search process, each identified common substructure is
evaluated by a scoring function and only substructures with the best
score are retained. The OEMCSSearch.SetMCSFunc
method provides an ability to set the scoring function of an
OEMCSSearch object, thereby influencing the result of
the maximum common substructure search process. See the
MCS scoring functions section for more information.
mcss.SetMCSFunc(oechem.OEMCSMaxAtoms())
The OEMCSSearch.Match method returns an iterator
over the maximum common substructures.
for count, match in enumerate(mcss.Match(target, unique)):
The OEMatchBase is then passed as an argument to the
OESubsetMol function, and subsequently converted
into a SMILES string.
oechem.OESubsetMol(m, match, True)
The detected maximum common substructure of the example program is
depicted in Figure: Example of MCSS. The following is the output
of the program in Listing 5:
Match 1:
pattern atoms: 0 1 2 3 4 5 6 7 8 9
target atoms: 7 5 3 4 1 2 0 8 9 10
match smiles = CCc1ccc(c(c1)O)O
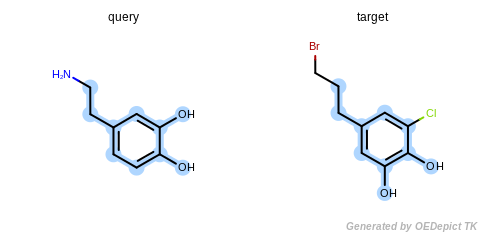
Example of MCSS
The maximum common substructure (highlighted by light blue) of dopamine and dopamine analog.
The maximum common substructure search can perform unique or
non-unique substructure searching by changing the second argument of
the OEMCSSearch.Match method. The default is a
non-unique search.
By definition, a match or subgraph is considered unique if it differs from all other subgraphs found previously by at least one atom or bond. Additionally, it is also considered unique if the pattern subgraph is mapped to a different part of the target.
Figure: Unique match shows an example in which the two matches are identified using the unique search method. Even though the two obtained subgraphs are identical, they represent different mappings between the pattern and the target, therefore they are both considered unique. Using a non-unique search would result in four matches, since the phenol can flip, yielding two additional matches.
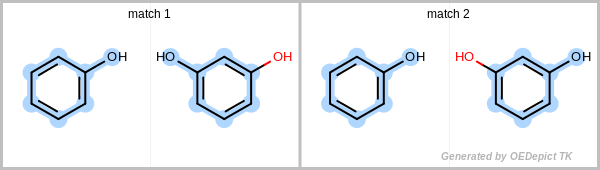
Unique match
Example for unique maximum common substructures
The search space of a maximum common subgraph determination can be
restricted by constraining pairs of atoms or bonds to be mapped onto
one another in all subgraph solutions. This is done using the
OEMCSSearch.AddConstraint method. Failure to
satisfy atom or bond pairwise constraints will prevent any subgraph
solutions from being identified. Constraints are considered satisfied
in subgraphs which do not contain any constrained atoms or bonds in
either the pattern or target molecules.
The AddConstraint method
returns true if a constraint is added successfully. If the pattern
atom or bond in the OEMatchPair does not exist as
part of the query molecule created in the initialization of the
OESubSearch object then
AddConstraint will
return false. Multiple calls to
AddConstraint using the
same pattern atom or bond will cause previously stored constraints to
be overwritten as constraints are mutually exclusive. It is impossible
to satisfy multiple simultaneous constraints for a single pattern atom
or bond, hence the exclusivity.
A read-only reference to the query molecule
OEQMolBase contained in an instance of
OEMCSSearch can be obtained with the
GetPattern method. Note
that if the OEMCSSearch was constructed with an
OEQMolBase, the returned OEQMolBase
is a separate object. OEQMolBase methods can then be
used to interrogate the returned OEQMolBase
reference.
The SetMaxMatches method
alters the maximum number of maximum common subgraph matches that will
be returned by the OEMCSSearch.Match method. The
search for maximum common substructures will not terminate immediately
upon reaching this limit. The maximum common subgraph cannot be known
unless the MCS is composed of all atoms and bonds of at least one of
the graphs being compared. The limit of subgraphs to be returned may
be reached with a smaller subgraph than the maximum. In such a case
the search continues for larger subgraphs until the search is
exhausted. OEMCSSearch.Match will return the first
N maximum common subgraphs where N is less than or equal to the
maximum match limit. The default limit set upon construction of an
OEMCSSearch instance is 1024 matches.
The SetMinAtoms method
sets the minimum number of atoms required of a subgraph match to be
returned by a MCS search. For example, changing the parameter of
SetMinAtoms in
Listing 5 to 11 would result in no solution
since there are only 10 atoms of the largest maximum common
substructure (see Figure: Example of MCSS).
mcss.SetMinAtoms(6)
A single atom can be a perfectly valid maximum common subgraph,
however, for many applications such a small subgraph may not be
considered useful. Setting the minimum number of atoms to a useful
size prevents unproductive subgraph matches from being returned by the
OEMCSSearch.Match method. The default set upon
construction of an OEMCSSearch instance for the
minimum number of atoms is one.
Exhaustive and approximate MCSS
The maximum common substructure search can be performed in two
different modes: a very fast method,
OEMCSType_Approximate, or a more comprehensive
method, OEMCSType_Exhaustive.
The type of the OEMCSSearch can be set at
initialization. The default value is
OEMCSType_Exhaustive.
The approximate method is based on traversing through pre-defined paths of the query structure and trying to map the visited query atoms into target atoms. Because these pre-defined paths represent only a fraction of all possible paths of a compound, it is not guaranteed that the approximate method can find the largest and all common substructures. Significant difference between the detected matches of the two methods could exist in cases when the query or target structure contains complex ring systems (fused or bridged) or stereo centers. However, comparing the two methods for thousands of structures revealed that these cases are rare and the approximate method provides a good trade-off between identifying MCS matches accurately and doing it 3 to 6 times faster than the exhaustive method.
Figure: Approximate MCSS and Figure: Exhaustive MCSS, show an example where the substructure identified by the approximate method is smaller by one atom then the solution identified by the exhaustive method.
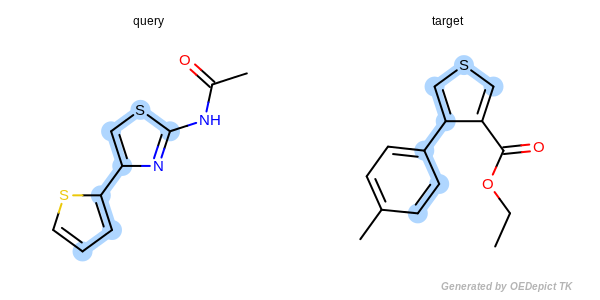
Approximate MCSS
Example for maximum common substructure identified by the approximate method.
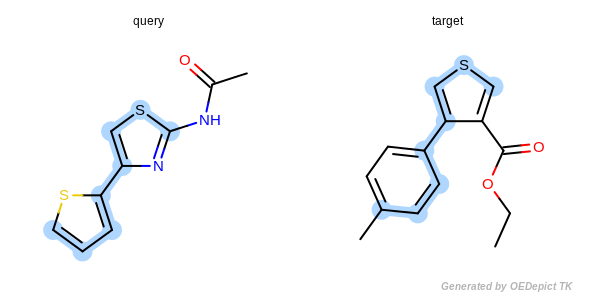
Exhaustive MCSS
Example for maximum common substructure identified by the exhaustive method.
Note
Using the approximate MCS is recommended if the speed of the search is crucial.
MCS scoring functions
OEMCSFunc is an abstract base-class that defines the
API used for scoring subgraph matches. The scores generated by
implementations of OEMCSFunc influence the sorting
and retention of maximum common subgraph matches generated by the
OEMCSSearch class. The scoring function is set by the
OEMCSSearch.SetMCSFunc method.
It is important to mention that using different scoring functions does not alter the way the search space is traversed to identify common substructures. It affects only how these identified substructures are evaluated.
Four implementations of the OEMCSFunc class are available in OEChem TK:
OEMCSMaxAtoms
The OEMCSMaxAtoms class is designed to order maximum common substructure matches by the maximum number of atoms included in the graph match. If two matches have the same number of atoms, then the tie is split based on the number of bonds contained in the match. (See example in Scoring A.)
Scoring function:
\(num.\ of\ mapped\ atoms + \frac{num.\ of\ mapped\ bonds}{100}\)
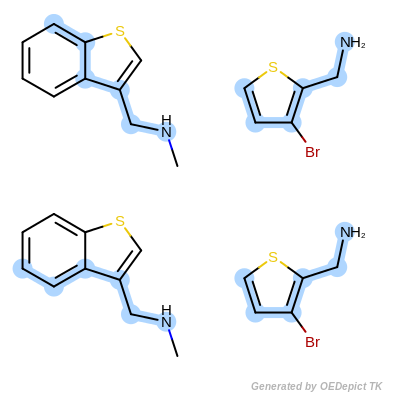
Scoring A
Retrieved matches using ‘OEMCSMaxAtoms’ as scoring function.
OEMCSMaxBonds
The OEMCSMaxBonds class is designed to
order maximum common substructure matches by the maximum number of bonds
included in the graph match.
If two matches have the same number of bonds, then the tie is split based on the
number of atoms contained in the match.
(See example in Scoring B.)
Scoring function:
\(num.\ of\ mapped\ bonds + \frac{num.\ of\ mapped\ atoms}{100}\)
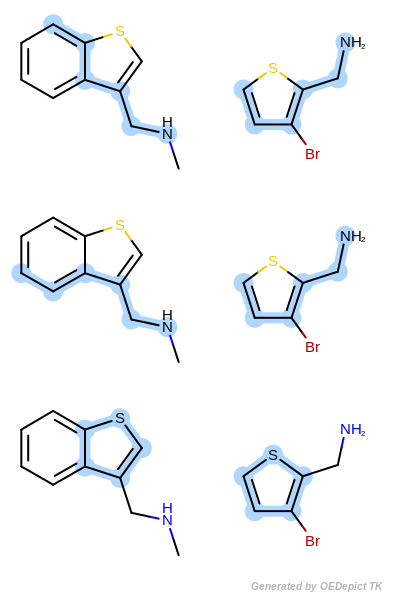
Scoring B
Retrieved matches using ‘OEMCSMaxBonds’ as scoring function.
OEMCSMaxAtomsCompleteCycles
The OEMCSMaxAtomsCompleteCycles
class is the same as the OEMCSMaxAtoms with
the addition of penalizing cyclic query bonds that are not mapped to any target
bonds, thereby giving priority to matches which contain complete cycles
common to both the pattern and the target structure.
(See example in Scoring C.)
Scoring function:
\(num.\ of\ mapped\ atoms + \frac{num.\ of\ mapped\ bonds}{100} - penalty \times num.\ of\ unmapped\ cyclic\ query\ bonds\)
The default penalty for each unmapped cyclic query bond is 1.0.
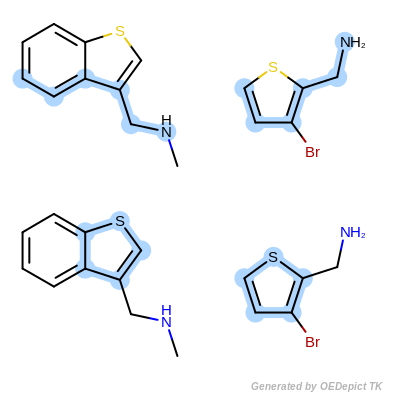
Scoring C
Retrieved matches using ‘OEMCSMaxAtomsCompleteCycles’ as scoring function.
OEMCSMaxBondsCompleteCycles
The OEMCSMaxBondsCompleteCycles class is the same as the OEMCSMaxBonds class with the addition of penalizing cyclic query bonds that are not mapped to any target bonds, thereby giving priority to matches which contain complete cycles common to both the pattern and the target structure. (See example in Scoring D.)
Scoring function:
\(num.\ of\ mapped\ bonds + \frac{num.\ of\ mapped\ atoms}{100} - penalty \times num.\ of\ unmapped\ cyclic\ query\ bonds\)
The default penalty for each unmapped cyclic query bond is 1.0.
Scoring D
Retrieved matches using ‘OEMCSMaxBondsCompleteCycles’ as scoring function.
It is important to remember that not only matches with the highest
score are retained, but also matches with scores higher than the best
score rounded down to the highest integer. In the example shown in
Scoring B three common substructures are detected
using the OEMCSMaxBonds scoring function. The first
two matches are scored 5.06, since they are composed of 5 mapped
bonds and 6 mapped atoms. There is only one other match which scored
higher than 5.0, this is the third retained match with a score of
5.05.
Clique Search
Clique detection is a bounded common substructure search. It is a useful search method in cases where common substructures other than the maximum common substructure need to be identified. The following example demonstrates a clique search.
Listing 6: Clique search example
from openeye import oechem
pattern = oechem.OEGraphMol()
target = oechem.OEGraphMol()
oechem.OESmilesToMol(pattern, "c1cc(O)c(O)cc1CCN")
oechem.OESmilesToMol(target, "c1c(O)c(O)c(Cl)cc1CCCBr")
# create clique search object
cs = oechem.OECliqueSearch(pattern, oechem.OEExprOpts_DefaultAtoms, oechem.OEExprOpts_DefaultBonds)
# ignore cliques that differ by more than 5 atoms from MCS
cs.SetSaveRange(5)
# loop over matches
for count, match in enumerate(cs.Match(target)):
print("Match %d :" % (count + 1))
print("pattern atoms:", end=" ")
for ma in match.GetAtoms():
print(ma.pattern.GetIdx(), end=" ")
print("\ntarget atoms: ", end=" ")
for ma in match.GetAtoms():
print(ma.target.GetIdx(), end=" ")
print()
The same molecules and expression options are used as in
Listing 5, however, an iterator over all
identified cliques is returned by the
OECliqueSearch.Match method. The
OECliqueSearch.SetSaveRange method bounds the
search. In this case, cliques returned will only differ by five nodes
relative to the maximum common substructure. The atom correspondences
for each of the returned cliques are printed in the example program.
OEExprOpts Namespace
Pattern matching in OEChem TK is always done using query molecules or
query graphs. Non-query molecules, i.e. those that are defined only
by OEMolBase abstract base-class must be converted
into a query molecule. Conversion into a query molecule is controlled
using the values in the OEExprOpts namespace.
Expression options can either be specified in the constructor for an
OEQMol, or using the convenience constructors in the
pattern matching classes, OESubSearch,
OEMCSSearch, OECliqueSearch which
take expression options as arguments.
Figure: Default example shows an example where the maximum common
substructure search is performed using the
OEExprOpts_DefaultAtoms and
OEExprOpts_DefaultBonds options.
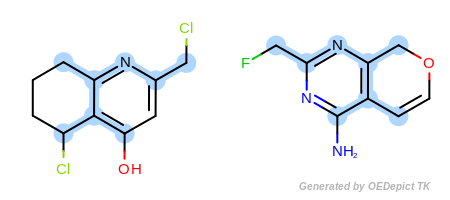
Default example
Example of maximum common substructure search with ‘DefaultAtoms’ and ‘DefaultBonds’ options
The OEExprOpts_DefaultAtoms option means that two
atoms are considered to be equivalent if they have the same atomic
number, aromaticity, and formal charge. The
OEExprOpts_DefaultBonds option means that two
bonds can be mapped to each other if they have the same bond order and
aromaticity.
Listing 7: MCSS with atom and bond expressions
from openeye import oechem
pattern = oechem.OEGraphMol()
target = oechem.OEGraphMol()
oechem.OESmilesToMol(pattern, "c1(cc(nc2c1C(CCC2)Cl)CCl)O")
oechem.OESmilesToMol(target, "c1(c2c(nc(n1)CF)COC=C2)N")
atomexpr = oechem.OEExprOpts_DefaultAtoms
bondexpr = oechem.OEExprOpts_DefaultBonds
patternQ = oechem.OEQMol(pattern)
# generate query with atom and bond expression options
patternQ.BuildExpressions(atomexpr, bondexpr)
mcss = oechem.OEMCSSearch(patternQ)
unique = True
count = 1
# loop over matches
for match in mcss.Match(target, unique):
print("Match %d:" % count)
print("Number of matched atoms: %d" % match.NumAtoms())
print("Number of matched bonds: %d" % match.NumBonds())
# create match subgraph
m = oechem.OEGraphMol()
oechem.OESubsetMol(m, match, True)
print("match smiles = %s" % oechem.OEMolToSmiles(m))
count += 1
The best way to understand how various atom and bond expressions
influence the pattern matching is to change the atom and bond
expressions in Listing 7 and compare the
obtained matches.
atomexpr = oechem.OEExprOpts_DefaultAtoms
bondexpr = oechem.OEExprOpts_DefaultBonds
After constructing the pattern molecule, the
BuildExpressions
method defines the level of atom and bond matching between the pattern
molecule and any target molecule.
patternQ.BuildExpressions(atomexpr, bondexpr)
By modifying the atom and bond expression options, very diverse pattern matching can be performed. Figure: Example A – Figure: Example E show several examples where maximum common substructure searches are performed for the same query and target molecules, but with various atom and bond expression options.
In the first example in Figure: Example A, the
OEExprOpts_ExactAtoms expression option is used to
give a higher degree of discrimination of the equivalence of atoms,
i.e. atoms can only be mapped to each other if they have the same
degree, number of hydrogens, chirality, mass, and ring membership, in
addition to the requirements of the
OEExprOpts_DefaultAtoms option.
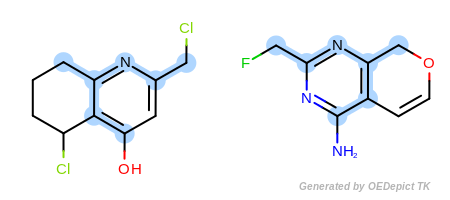
Example A
`ExactAtoms` and ‘DefaultBonds`
Figure: Example B – Figure: Example E
show examples where the
discrimination capability of the
OEExprOpts_DefaultAtoms is decreased by adding
various modifiers. For example, using the
OEExprOpts_EqAromatic modifier, atoms in any
aromatic ring systems are considered equivalent. As a result, the
pyridine and pyrimidine ring can be mapped to each other in
Figure: Example B. Similarly,
OEExprOpts_EqHalogen (Figure: Example C) and
OEExprOpts_EqONS (Figure: Example D) define
equivalency between halogen atoms and oxygen-nitrogen-sulfur atoms,
respectively. Using OEExprOpts_EqCAliphaticONS
(Figure: Example E) an aliphatic query carbon atom is considered
equivalent to any oxygen, nitrogen, or sulfur atom.
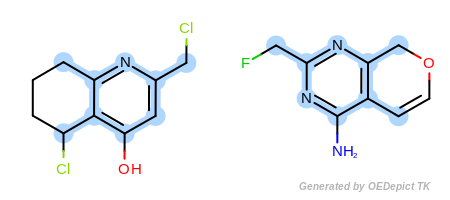
Example B
‘DefaultAtoms|EqAromatic’ and ‘DefaultBonds’
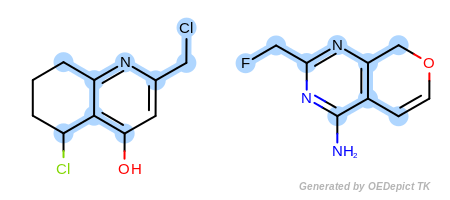
Example C
‘DefaultAtoms|EqHalogen’ and ‘DefaultBonds’
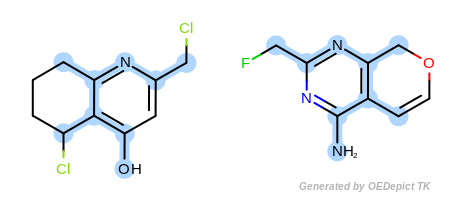
Example D
‘DefaultAtoms|EqONS’ and ‘DefaultBonds’
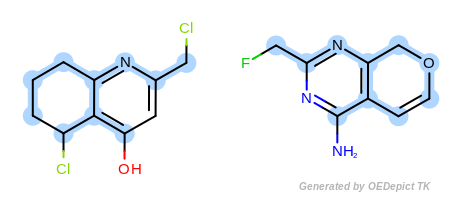
Example E
‘DefaultAtoms|EqCAliphaticONS’ and ‘DefaultBonds’
Similar modifiers exist for altering bond equivalency.
Figure: Example F shows an example where single and double bonds
are considered identical when
OEExprOpts_EqSingleDouble modifier is utilized.
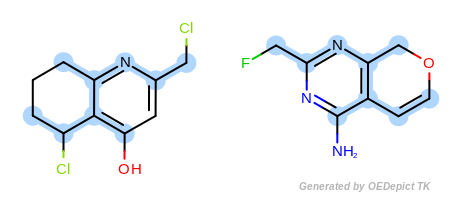
Example F
‘DefaultAtoms’ and ‘DefaultBonds|EqSingleDouble’
The last example in Figure: Example G represents a very unrestrained search, where both the atom and bond expression options have weak discrimination power.
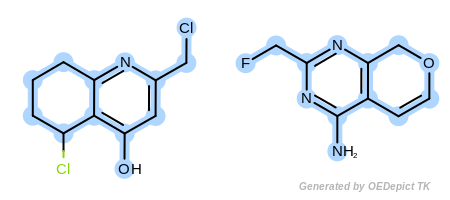
Example G
‘DefaultAtoms|EqAromatic|EqCAliphaticONS|EqHalogen|EqONS’ and ‘DefaultBonds|EqSingleDouble’
Even though only maximum common substructure search examples are
presented here, atom and bond expression options can be similarly used
with substructure searches or clique detections. For a full
description of expression options and their usage please refer to the
OEExprOpts namespace section in OEChem TK API.