UserInertialStarts¶
OEFastROCSOrientation.UserInertialStarts allows custom
coordinates to be used as starting positions for optimization. Using the
4TMN ligand from the
Introduction as an example,
optimization can be done at the atom highlighted in green below:
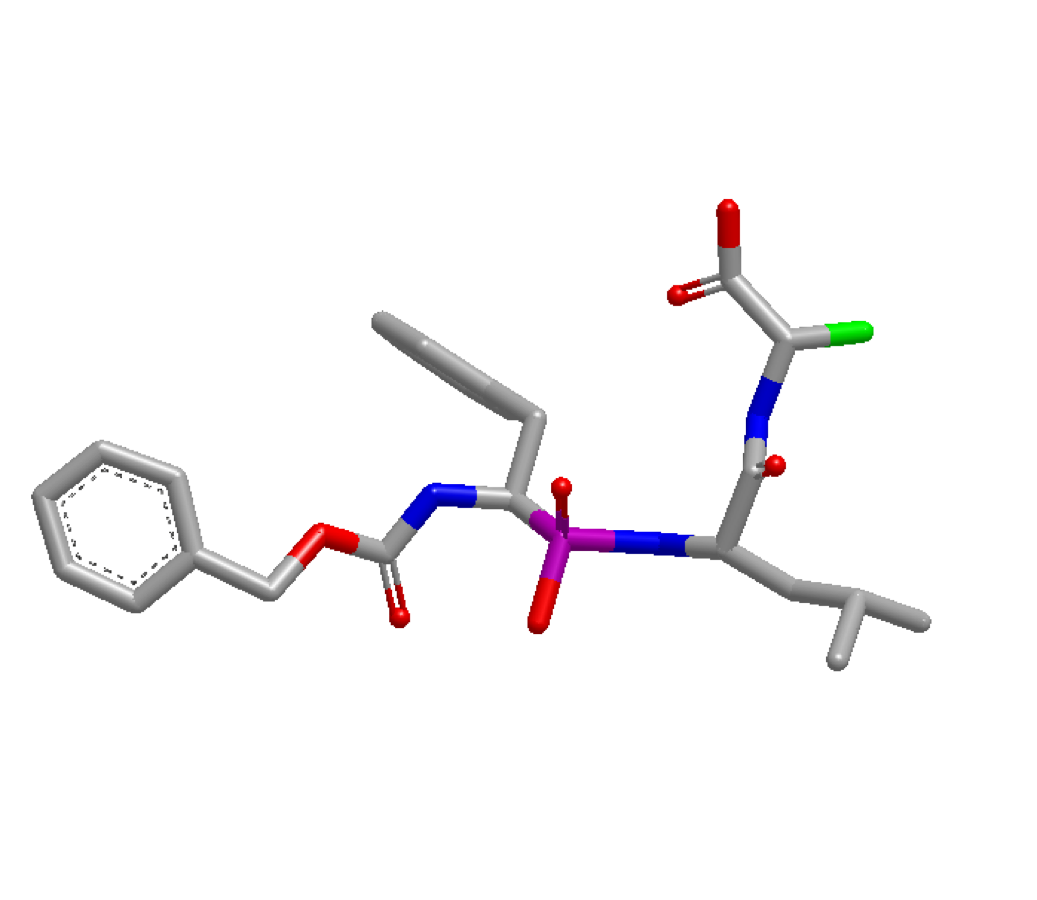
4TMN Ligand with atom 34 highlighted green¶
Before running the program, the index of the desired atom needs to be identified. This can be accomplished by loading the molecule in VIDA and turning on atom indices. In this example, the atom’s index is identified as idx 34. With this information, the x,y,z coordinates of atom idx 34 are pulled via the code snippet below. These coordinates are then used as user-defined starting coordinates: