FLYNN Usage¶
Overview¶
Fitting small molecules to electron density is a challenging problem. Small molecules with few rotatable bonds are amenable to hand placement in density. Add a few rotatable bonds, however, and the problem becomes factorially more complicated. Furthermore, unlike protein modeling, many small molecules are relatively unique entities whose conformation cannot be predicted from previous homologies.
FLYNN uses a high-quality conformational analysis and a fast rigid overlay to position ligands in density in conjunction with a combined forcefield to further position these conformations while, at the same time, minimizing ligand strain. FLYNN can automatically locate ligand density and automatically fit fragment cocktails, or, failing that, FLYNN can use a supplied bounding box in which to place the ligands of interest.
Ligands are fit to density, however the placements are scored and ranked based on the real space correlation coefficient (RSCC) and optionally protein based docking scores: Piecewise-Linear-Potential (PLP) and Chemscore.
FLYNN reads common map formats such as CCP4, CNX/CNS (XPLOR) and MTZ as well as OpenEye’s grid formats and a plethora of small molecule, protein and connection table formats.
Minimum Requirements¶
At the minimum, FLYNN requires a density file and a the connection table of the ligand - usually derived from a supplied molecule file e.g. .pdb, .sdf, .mol2, .smi. FLYNN automatically searches the density for volumes of unmodeled density that are similar in size to the input ligand. Because of this it is highly recommended that a protein is also input to FLYNN. This makes the job of locating the ligand density less prone to false positives.
Based on analyzing several hundred protein/ligand combinations, the best way to use FLYNN for ligand fitting is described as follows (of course your mileage may vary):
Always use a protein:
The protein is used to identify modeled density where the ligand cannot be placed. Without this, FLYNN will identify many candidate locations for placing the ligand that will need to be analyzed.
Use difference maps when appropriate:
The reflection data should be refined without a previous ligand conformation or generated without the ligand present. While in many cases the same result will be generated, if the ligand is fit to data refined with a previous ligand conformation, FLYNN may be biased towards the density modified by the pre-existing ligand conformation.
use fragment mode for fitting fragments
If you have an input cocktail, use the -fragment flag. This option simultaneously fits all the cocktails against density and sorts them by best fit.
Types of input ligands¶
Not all ligands can be successfully fit by FLYNN. Such types include large polypeptides or proteins, very flexible molecules, or simply molecules that contain atoms that are not present in the MMFF94s force-field.
Perhaps the most important property is rotatable bond count. Although FLYNN may be able to generate conformers for molecules with more than 20 rotatable bonds, the results of such an exercise would be dubious at best and should be taken with a grain of salt.
FLYNN will automatically check for unhandled atom types, but will gladly try and fit large molecules or proteins that contain many rotatable bonds usually with disappointing results.
Note
Dangers of PDB format for ligand input Many crystallography packages do not enforce writing out connection table or bond order information when outputting pdb files, or worse, strip them out. If bond distances are not correct FLYNN’s attempt to automatically assign bond orders and atom types can be suspect. To facilitate this, FLYNN includes a -precheck flag that can be used to verify that the input ligand is identified correctly and for validation of the automatic bond order assignments.
Stereochemistry Enumeration¶
Compounds that contain unspecified or ambiguous definitions of stereochemistry will be preprocessed before conformational sampling to explicitly enumerate stereochemistry. Input molecules that have three dimensional coordinates inherently have stereochemistry specified, but SMILES or two dimensional SD files may have atoms (R/S) or bonds (E/Z) for which the stereochemistry is unknown or unspecified. In the case where the stereochemistry supplied is suspect, FLYNN provides an option that will completely enumerate all stereochemical centers.
The resulting ligands will be sorted based on their best fit to density regardless of the stereochemistry. That is, the best solutions will percolate to the top of the output. See Input Options below for more details.
Refinement Dictionaries¶
Both FLYNN and its companion program WRITEDICT can generate refinement dictionaries. A refinement dictionary is needed to maintain proper ligand geometry when using external reciprocal space refinement programs such as REFMAC5 or CNS X/PLOR.
Refinement dictionaries are a list of geometrical constraints encoding used chemical bonding and molecular conformation information. They are used by various refinement packages to describe standard geometries and constraints that are used during the refinement process. The quality of the post refinement ligand conformations are directly related to the quality of the constraints [AFITT-CL Vagin-2004]_.
FLYNN comes bundled with a refinement dictionary writer named WRITEDICT. WRITEDICT uses the MMFF94 forcefield to derive geometrical constraints for the input ligands or ligand-protein complexes. The output dictionaries enforce, as closely as possible, the input ligand’s geometries while allowing the refinement programs to modify the geometry as needed.
WRITEDICT also automatically detects covalent bonds in pdb files and inserts the appropriate PDB LINK records and covalent bond entries in the output refinement dictionary.
Note about REFMAC5 refinement dictionaries¶
When outputting REFMAC5 refinement dictionaries, WRITEDICT writes out a .cif file and a .pdb file. The .pdb file is written out for two reasons:
Inconsistent Hydrogen Naming
Some applications write out hydrogen names incorrectly in ways that cause REFMAC5 or visualization programs like coot or WinCoot to be unable to associate the hydrogens in the refinement dictionary with hydrogens in the pdb file. In the worst case, REFMAC5 will crash entirely.
When necessary, WRITEDICT renames and renumbers hydrogens, if they exist, so that REFMAC5 won’t crash and so the .cif file is consistent with the .pdb file.
Covalent Bonds
WRITEDICT detects covalent bonds and outputs proper LINK records in the .pdb file and proper covalent constraints in the .cif file. Without using both outputs, REFMAC5 will not detect covalent bonds during refinement.
Known residues
When WRITEDICT detects a known residue, it may remap the atom names to be canonical with the known residue (this prevents REFMAC5 from failing to refine). These new atom names are saved in the .pdb file that WRITEDICT outputs.
Input Ligands with no residue information
When WRITEDICT analyzes a ligand with no residue information, it assigns the whole ligand to a single residue (by default UNL). If this is not the desired outcome, the ligand must be put into PDB or MOL2 format and the residues must be manually assigned.
It is highly recommended to use WRITEDICT’s generated .pdb file in conjunction with the .cif.
Looking up known residues¶
WRITEDICT has an internal dictionary of known residues. By default, known residues names are retained when the graph of the known residue is exactly the same as the input residue. When this occurs, writedict can replace REFMAC5’s dictionary with the MMFF94 generated dictionary. If the graphs do not match then WRITEDICT relabels the residue with a new residue name since a different dictionary needs to be created.
Options available for residue matching is as follows:
Option Meaning exact Known residue and input residue graphs must match exactly. substructure Input residue may be a substructure of the known residue.
Cannot be used with the exact flag.
fuzzybonds Bond orders do not need to match atomname Atom names must match
Options are set using the -strict flag and joined with colons (:).
The default is:
-strict substructure:fuzzybonds
To reject dictionaries that do not match exactly, use:
-strict exact
However, the more permissive default setting has been known to generate invalid dictionaries on occasion. It is always safe to force exact matches.
To have WRITEDICT attempt to see if the ligand’s residue has already been deposited in RCSB simply add the -lookup switch. This forces WRITEDICT to compare (based on the current -strict settings) the input residue to all known residues. This is useful if the input is from a 2D connection table, such as smiles or does not contain residue information.
Note on the MMFF94 versus MMFF94s forcefields¶
The MMFF94s forcefield is a variant of MMFF94 that emulates time-averaged structures typically observed during crystallographic structure determination, mainly planar geometries at unstrained delocalized trigonal nitrogen centers. However, there are many theoretical studies that show nitrogen centers are puckered [AFITT-CL Halgren-VI-1999]_.
That being said, due to the prevalence of crystallographic examples where time averaging has occurred, many chemists erroneously consider the time-averaged structure to be correct, hence this variant is available for use.
WRITEDICT approximates the MMFF94s forcefield by enforcing planar aniline nitrogen configurations using the out of plane atom types and parameters from the MMFF94s. See the -planarAniline parameter to specify planar constraints.
Note on FLYNN and writing CIF Files¶
When FLYNN is run, CIF dictionary files are always written as output. Because the dictionary files are not conformation independent, they are written as if the molecules were split into separate files whether or not the user specifies FLYNN’s -split flag. (See the split flag in Advanced Parameters below for more details). These files are numbered, however, so that the file with the lowest number corresponds to the best scored fit ligand.
For example, consider using the -sortBy plp flag and getting the following output:
1nhu-poses_n001_b001_s01_c003.pdb 1nhu-poses_n002_b001_s01_c000.pdb 1nhu-poses_n003_b001_s01_c001.pdb 1nhu-poses_n004_b001_s01_c002.pdb
This indicates that the best fit to density (c000) has the second best PLP score (n001). By default the files are sorted by RSCC.
FLYNN CIF output consists of the following files:
- a CIF file
- a PDB file (in case atom names were remapped) or links were added.
- an OEB file that contains the original molecule with annotations
It is safest, however, to run WRITEDICT on the output ligand and protein complex. The CIF files generated by FLYNN are primarily useful for adjusting torsions and geometries in various ligand building programs, such as coot.
Command Line Interface¶
A description of the command line interface can be obtained by executing FLYNN with the --help option.
> flynn --help
will generate the following output:
Help functions:
flynn --help simple : Get a list of simple parameters
flynn --help all : Get a complete list of parameters
flynn --help <parameter> : Get detailed help on a parameter
flynn --help html : Create an html help file for this program
Required Parameters¶
- -in <filename>¶
File containing a molecule to be fit to density.
File type Extension SMILES .smi .ism .smi.gz .ism.gz OEBinary .oeb .oeb.gz SDF .sdf .mol .sdf.gz .mol.gz MOL2 .mol2 .mol2.gz PDB .pdb .ent .pdb.gz .ent.gz MacroModel .mmod .mmod.gz
- -out <filename>¶
File containing resulting conformations exported in the following formats:
File type Extension OEBinary .oeb .oeb.gz SDF .sdf .mol .sdf.gz .mol.gz MOL2 .mol2 .mol2.gz PDB .pdb .ent .pdb.gz .ent.gz MacroModel .mmod .mmod.gz
- -map <filename>¶
Input density grid used to fit ligand. Note that mtz files have additional settings, see MTZ File Parameters
Grid File type Extension MTZ .mtz OpenEye .grd Grasp .phi CCP4 .map .ccp4 XPLOR .xplor xplmap ASCII Grid .agd
Optional Parameters¶
Input Options¶
- -prot <filename>¶
Optional protein used to mask density. This model is used to mask away density where the ligand should not be placed. While the protein is not required, it is highly recommended.
File type Extension OEBinary .oeb .oeb.gz SDF .sdf .mol .sdf.gz .mol.gz MOL2 .mol2 .mol2.gz PDB .pdb .ent .pdb.gz .ent.gz MacroModel .mmod .mmod.gz
- -fragment¶
Fragments are fit taking the input fragment cocktail and, one at a time, fitting each fragment against each region of detected density. Once a fragment has been placed, it is further analyzed to ensure that all possible orientations of the fragment have been sampled. In poor density, several orientations may fit equally well. To break ties, FLYNN scores each pose with the following scores:
o RSCC (real space correlation coefficient) This is a measure of fit to electron density. [AFITT-CL Jones-1991]_
o PLP Piecewise-linear potential. [AFITT-CL Verkhivker-2000]_
o Chemscore [AFITT-CL Eldridge-1997]_
The docking scores are not used to fit the molecule, they are only used to rank the output. Unless highly symmetric molecules are being input, the real space correlation coefficient (RSCC) is the preferred method of ranking results to density.
All scores are annotated to the output ligands using SD data when appropriate (.SD and .OEB output) or using the PDB remark field:
Example PDB output
REMARK OpenEye Flynn MMFF/Shape v 2.1.0
REMARK Stereo Variant: 1
REMARK Blob: 1
REMARK Conformer: 1
REMARK Tanimoto Shape: 0.2004
REMARK Tanimoto MMFF/Shape: 0.2034
REMARK Local Strain: 4.9316
REMARK RSCC: 0.529438
REMARK PLP: -51.395416
REMARK Chemscore: -16.288113
Example SDF output
> <Tanimoto MMFF/SHAPE>
0.2034
> <Tanimoto Shape>
0.2004
> <Overlap>
364.767659122018
> <Fit Overlap>
781.301114117834
> <Local Strain>
4.9316
> <Lambda>
1.600000023842
> <Ref Overlap>
1371.829467773438
> <Stereo>
1
> <Blob>
1
> <Conformer>
1
> <RSCC>
0.529438
> <PLP>
-51.395416
> <Chemscore>
-16.288113
To use fragment mode, please add the “-fragment” option to the command line. In future versions of FLYNN, this will most likely become the default setting.
The output of the fragment fitting process is a file for each density region that includes the fragments fit to the region sorted from best-fit to worst fit. For example:
> flynn -in fragments.smi -out 2IKO_cocktail.sdf ...
would result in the files:
2IKO_cocktail_blob001.sdf
2IKO_cocktail_blob002.sdf
2IKO_cocktail_blob003.sdf
one for each blob found. The first molecule in the file is the best fit to the density.
To sort using another measure, for instance, PLP, use the sort flag:
> flynn -sortBy plp -in fragments.smi -out 2IKO_cocktail.sdf ...
When oeb format is used, the blob is also stored in the OEB file. When using VIDA, simply expand the molecule to investigate the blobs density.
- -param <parameter filename>¶
A parameter file is a text file that lists parameter settings to be used during a run. If a parameter is specified both on the command line and in the parameter file, the value specified on the command line is used.
The format of the parameter file is as follows:
- One parameter per line
- For non-list parameters one key-value pair per line.
- For list parameters a key followed by all the values.
- Boolean parameters must be listed as a key followed by true or false.
- The parameter file may not contain the -param parameter.
- Lines begining with # are considered comments
- -prefix <file prefix>¶
Define the default prefix to use for loggging and report file generation.
This -prefix is overridden when using the -reporthtml or -reportfile command line options.
[default=flynn]
- -manualSearch <filename>¶
Use the input molecule to generate a box volume. All density inside this box will be used to place the ligand.
- -box <filename>¶
Alias for -manualSearch
- -boxpad <number>¶
Pad the box created with the -manualSearch flag by an amount in Ångströms.
[default=0.0]
- -autoThenManualSearch¶
Automatically search for blobs first, if now suitable blobs are found, use the supplied box to bound density.
[default=false]
- -blobsThenBox¶
- -densityAsIs¶
Use the supplied density as it is; do not try to find suitable blobs inside the density. Note that the supplied density has to be pretty close to the actual ligand density for this to work
[default=false]
- -distance <value>¶
Reject blobs whose average distance to the protein is greater than this value.
[default=4.0]
- -ligandAsIs¶
Do not generate conformations for the ligand; use only the supplied conformations.
- -mmff94s¶
- Use the MMFF94s variant of the MMFF94 forcefield ( this uses planar aniline nitrogens ).
[default=false]
- -reportfile <filename>¶
- File location for writing the report file. This is a comma separated file containing specifics about the results.
Smiles,Blob,Stereo Variant,Conformer,Tanimoto Shape,Tanimoto MMFF/SHAPE,Local Strain CC(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CCC4=CC(=O)CC[C@]34C)C,1,,,0.4110,0.4296,1.342 CC(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CCC4=CC(=O)CC[C@]34C)C,1,,,0.313,0.3704,4.553 CC(=O)[C@H]1CC[C@@H]2[C@@]1(CC[C@H]3[C@H]2CCC4=CC(=O)CC[C@]34C)C,1,,,0.3265,0.3628,5.223
- -resname <name>¶
Set the output residue name to <name>. <name> must be less than or equal to three characters in length. This forces the output residue to be named <name> even if the residues structure does not match a known deposited residue.
- -chainid <chainid>¶
Forces the ligand to be placed in the chain <chainid>
- -sortAllChiral <true/false>¶
If set to true, when multiple chiralities are enumerated, sort all chiralities from best to worst per blob, otherwise sort chiral structures independently. For example, if two blobs are found, then the output will have all chiral structures sorted from best fit to worst fit for the first blob, and then all chiral structures sorted from best fit to worst fit for the second blobs, etc Note that the second blob may contain better fits than the first. (Note: This may interleave chiral structures).
[default=true]
- -verbose¶
- This is a boolean flag that controls the level of
detail written to the log file. By default FLYNN will only write minimal information to the console. Verbose logging will cause more information to be written to the log file in order to follow behavior during program execution.
[default=false]
MTZ File Parameters¶
- -autoMTZ¶
Automatically try to open the mtz file using the DELWT and FDELWT columns from REFMAC5 mtz files.
[default=true]
- -Fc <columnname>¶
Column to use for Fc. Note, to load arbitrary columns, you can use the *-Fc** and -Phic columns and the Fc maptype.*
[default=FC]
- -Fdelwt <columnname>¶
Column to use for Fdelwt or difference map amplitudes
[default=F]
- -Fobs <columnname>¶
Column to use for FObs.
[default=F]
- -Fwt <columnname>¶
Column to use for Fwt or regular map amplitudes.
[default=FWT]
- -Phic <columnname>¶
Column to use for Phic.
[default=PHIC]
- -Phidelwt <columnname>¶
Column to use for Phdelwt or difference map phases.
- -Phiwt <columnname>¶
Column to use for Phwt or regular map phases.
- -mtype¶
- The map type to use for fitting. Fo-Fc, Fc, 2Fo-Fc, 3Fo-Fc, Fwt,
- Fdelwt...
[default=2Fo-Fc]
Advanced Parameters¶
- -ciftype¶
Specify the format of the refinement dictionary. Legal values for refinement dictionaries are ‘refmac’, ‘phenix’ and ‘buster’. Refinement dictionaries are always written out using the designation specified in the ‘-split’ command. For best results, use -split when writing out cif files when using FLYNN.
- -flipper¶
Force enumeration of all stereochemical centers. Otherwise, only missing stereo chemistry will be enumerated.
[default=false]
- -overlays¶
Set the number of initial rigidly fit overlays to optimize into density.
[default=10]
- -precheck¶
Perform only a preliminary check of the data to see if fitting is possible. (This flag is meaningless without the -reporthtml flag).
[default=false]
- -reporthtml¶
Generate an html report of the fitting process. This is a useful first step to verify the data. It also includes a 2D image of the ligand that is being fit in order to verify bond orders. All output of FLYNN will be captured in the specified html file.
- -residues¶
Comma separated list of residues to use as distance constraints. If residues are specified, the -distance flag will use the residues to reject blobs that are too far away. Note that with a residue list, the distance computed is a minimum distance, not an average distance. Residues must be specified as <residue number><chain id>. For example: 120C,134C. On Windows, double quotes must be added to specify two or more residues, i.e. -residues “120C,134C”.
- -rms¶
If the input ligand has 3D coordinates, this calculates the RMS distance to the ligand and records it in the molecule. If -split is on, it also records it in the file name.
[default=false]
- -split¶
Split the result into different files. Each filename is annotated with the resulting shape score so that when the output directory is listed alphabetically, the best scores will be displayed in order. In the case where the output format is pdb the ligand fitting results are written in REMARK statements at the top of the file.
The splits are labeled as follows outputname_n###_b###_s###_c### where n### is the number of the ordered results, b### is the blob for the result, s### is the stereovariant and c### is the number of the conformer fit as ordered by MMFF/Shape.
[default=false]
- -suppressH¶
By default FLYNN outputs hydrogens of the resulting poses. Setting this to true will not output hydrogens. It is always recommended to output hydrogrens for ligands in order to keep their chemistry the same as flynn’s internal optimizer. Only suppress hydrogens if your refinement package doesn’t handle hydrogen names correctly.
[default=false]
- -fix_serial_numbers¶
Force serial numbers to be unique when writing out the resulting pdb file for use in the refinement package. This is usually caused by adding hydrogens to the ligand outside of the context of the protein and then using it in conjunction with the original PDB structure.
[default=false]
Fragment Fitting Parameters¶
- -blobTanimoto¶
Density is searched for each input ligand. This flag sets the minimum tanimoto overlap for which to consider two blobs the same. When pruning similar blobs, the larger is taken.
[default = 0.45]
- -sortBy¶
Choose the fragment sorting function, tanimoto, rscc (real space correlation coefficient), plp (piecewise linear potential) or chemscore.
The fragments are fit in the normal fashion (tanimoto), but then re-scored and sorted using the chosen metric.
[default = rscc]
3D Construction Parameters and Torsion Driving Parameters¶
These are advanced conformer generation parameters. Normally, they do not need to be adjusted. Please consult the OMEGA manual for assistance:
- -ewindow¶
Controls the MMFF energy window when generating initial conformations. Normally, this setting does not need to be adjusted, however in some cases large floppy molcules may require an adjustment. This setting is in kcals/mol.
[default = 15.0]
Example Commands¶
The example commands in this section can be run with files found in the data/1nhu directory under the top-level installation directory.
Basic Usage¶
> flynn -in 1nhu.ism -map 1nhu_prot.mtz -prot 1nhu_prot.pdb \
-out 1nhu-poses.pdb
Use the ligand specified by 1nhu.ism and fit it against the density in the mtz file 1nhu_prot.mtz. Since this is an mtz file and -autoMTZ is defaulting to true, the Fdelwt map will automatically be generated and used as the density target. The protein atoms specified in 1nhu_prot.pdb will be used to mask the density before the map is searched for appropriately sized ligand volumes.
Generating Reports¶
> flynn -in 1nhu.ism -map 1nhu_prot.mtz -prot 1nhu_prot.pdb \
-out 1nhu-poses.pdb -precheck -reporthtml 1nhu.html
Performs a preliminary check of the data, ensuring that the ligand can be processed and that the mtz file can be opened. An html file is generated 1nhu.html with a corresponding image file 1nhu.html.gif. The html file can be opened with a web browser to view the ligand being fit and any corresponding issues.
The output will be written to 1nhu-poses.pdb in pdb format.
Choosing box regions of density¶
> flynn -in 1nhu.ism -map 1nhu_sigma.mtz -prot 1nhu_prot.ent \
-out 1nhu_poses.pdb -box 1nhu.pdb -boxpad 2.0
This is the same as above except that the density is pruned down to the bounding box specified by 1nhu.pdb padded by 2.0 Ångströms. Furthermore, if a suitable blob is not found, then the pruned density will be used as is. This is quite effective for regions of density with a high degrees of disorder. It is also useful to select this option when using the supplied interface to coot.
Choosing density close to residues¶
Use the -residues and -distance flags to indicate residues close near the desired density. Only blobs that are found within -distance to the selected residues will be selected. Residues are specified by <residuenumber><chain> separated by commas.
> flynn -in 1nhu.ism -map 1nhu_prot.mtz -prot 1nhu_prot.pdb \
-out 1nhu-poses.pdb -residues 528B,477B
Choosing MTZ columns¶
> flynn -in 1nhu.ism -map 1nhu_sigma.mtz -prot 1nhu_prot.ent \
-out 1nhu_poses.pdb -box 1nhu.pdb -boxpad 2.0 \
-boxThenBlobs -autoMTZ false -Fobs Frefined1 -Fc FC \
-Phic PHIC -mtype 2Fo-Fc -autoMTZ false
This example shows how to choose a non-standard column from the MTZ file. the map is generated using Frefined1 as the observed amplitude column, FC as the calculated amplitude column and PHIC as the calculated phase column. Furthermore, the regular map 2Fo-Fc is used. Note that -autoMTZ must be set to false for these settings to take effect.
Fragment usage¶
The -fragment flag may be included with any of the above commands. There are three main differences when running in fragment mode:
- Multiple ligands are fit simultaneously.
- If the blobs detected for difference ligands overlap, the ligands are assigned to the same blob. (This is for reporting purposes)
- Ligands are sorted according to the Real Space Correlated Coefficient.
> flynn -fragment -in 1nhu.ism -map 1nhu_prot.mtz -prot 1nhu_prot.pdb \
-out 1nhu-poses.sdf
When in fragment mode, it is better to output to a format that supports SD style data (.sdf, .oeb). This makes it much easier to analyze the results in a molecular viewer that supports a sortable spreadsheet. (VIDA, MOE and so on)
To sort the results using another scoring function, use the -sortBy flag.
> flynn -sortBy chemscore -in 1nhu.smi ...
Results on The Gold Test Set¶
Included in the FLYNN distribution is a copy of the protein$+$ligand complexes that have structures factors available from the Electron Density Server [AFITT-CL EDS]_.
Along with this data set is a simple python script that can be used to fit ligands to the test data set. OpenEye has prepared the input test set as follows:
- The Structure Factors and protein complexes were downloaded from EDS.
- The ligands were separated from the proteins.
- The protein was rerefined using the original mtz file to remove ligand bias.
A simple directory structure was created as follows:
- pdbcode/protein.pdb - the re-refined protein
- pdbcode/ligand.pdb - the original ligand
- pdbcode/protein.mtz - the re-refined difference map density.
The following python script was used to run FLYNN on all of the proteins. (This script is also included in the FLYNN distribution):
#########################################################################################
## Copyright (C) OpenEye Scientific 2007,2008,2009,2010,2011
#########################################################################################
## This script analyzes the Gold dataset shipped with the flynn distribution
import os,sys
command = "../../../bin/flynn -in %(CODE)s/ligand.pdb -map %(CODE)s/protein.mtz " \
"-prot %(CODE)s/protein.pdb -out %(CODE)s_results.sdf " \
"-verbose -rms -reportfile %(CODE)s.log %(REDIRECT)s %(CODE)s.out"
if sys.platform == "win32":
REDIRECT = "2>"
else:
REDIRECT = "&>"
RMS = []
for file in os.listdir("."):
print file
if os.path.isdir(file) and len(file) == 4:
code = file
CMD = command % {"CODE":code, "REDIRECT": REDIRECT}
print CMD
os.system(CMD)
index = None
rmses = []
for line in open("%s.log"%code):
if index is None:
index = line.strip().split(",").index("RMSD")
else:
rmses.append( float(line.strip().split(",")[index]) )
if rmses:
print "\t===>Lowest RMS", min(rmses)
The results of FLYNN are shown in Figure Gold Test Set Results. The mean RMS to the crystal structure is 0.676 Angstroms.
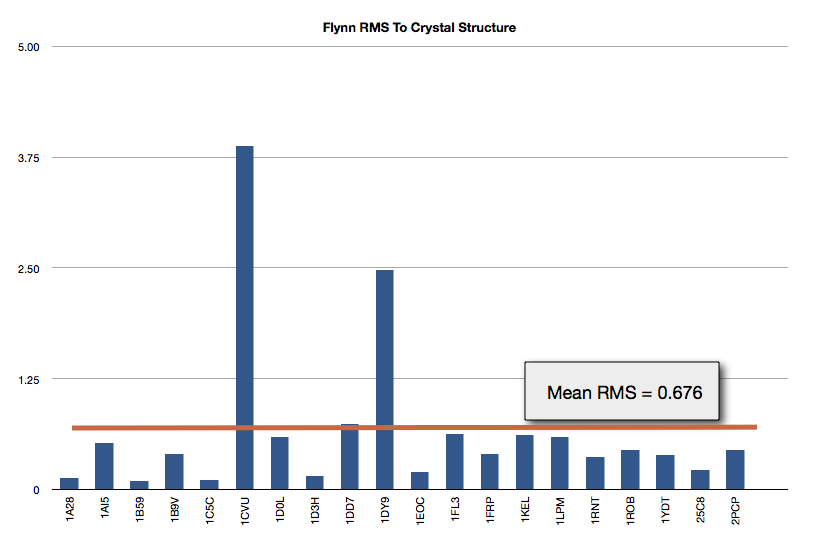
Gold Test Set Results
WRITEDICT Usage¶
Command Line Interface¶
A description of the command line interface can be obtained by executing WRITEDICT with the --help option.
> writedict --help
will generate the following output:
Help functions:
writedict --help simple : Get a list of simple parameters
writedict --help all : Get a complete list of parameters
writedict --help <parameter> : Get detailed help on a parameter
writedict --help html : Create an html help file for this program
Required Parameters¶
- -in <filename>¶
File containing a molecule to be fit to density.
File type Extension SMILES .smi .ism .smi.gz .ism.gz OEBinary .oeb .oeb.gz SDF .sdf .mol .sdf.gz .mol.gz MOL2 .mol2 .mol2.gz PDB .pdb .ent .pdb.gz .ent.gz MacroModel .mmod .mmod.gz
- -out <filename prefix>¶
This specifies a prefix for outputting the refinement dictionary .cif or .xplor and the pdb file. The pdb file ( and pdb.oeb) is always output and should be used for purposes of refinement since WRITEDICT sometimes corrects faulty hydrogen atom names and will include LINK records in the pdb if covalent bonds are detected.
The log file will also be written using this prefix as <filename prefix>.log.
- -prefix <filename prefix>¶
Alias for -out
Output Options¶
- -assignCCP4¶
If true then CCP4 atom types will be assigned to the output pdb file.
[default=true]
- -includeRotors¶
Set to true if the output should include rotor torsion terms. If this is set to false, refinement programs will not be able to modify any rotors.
[default=true]
- -suppressH¶
If true, no hydrogens are output.
[default=false]
- -type¶
set this to 0 for REFMAC5 refinement dictionaries or 1 for CNS/CNX (XPLOR). By default REFMAC5 dictionaries are generated.
[default=0]
- -planarAniline¶
Use the MMFF94s variant of the MMFF94 forcefield. This forcefield emulates time-averages structures observed in crystallographic and other structure determination methods.
The MMFF94s forcefield is a variant of MMFF94 that emulates time-averages structures typically observed during crystallographic structure determination, mainly planar geometries at unstrained delocalized trigonal nitrogen centers. However, there are many theoretical studies that show puckering at nitrogen centers. (see MMFF VI. in JCICS)
That being said, due to the prevalence of crystallographic studies, many chemist erroneously consider the time-averaged structure to be correct, hence this variant is available for use.
[default = false]
- -strict¶
Change the known residue matching mode.
Options available for residue matching is as follows:
Option Meaning exact Known residue and input residue graphs must match exactly. substructure Input residue may be a substructure of the known residue.
Cannot be used with the exact flag.
fuzzybonds Bond orders do not need to match atomname Atom names must match Options are separated by colons. The default settings are set to limit the number of REFMAC5 issues. -strict exact:substructure will find more matches, but increases the risk of REFMAC5 rejecting the dictionary.
[default = exact:fuzzybonds]
- -lookup¶
Attempts to lookup residues from the internal dictionary store using the current -strict status. This is useful to find out if a residue has already been deposited in the RCSB.
If -lookup is not set, mismatching residues will be given new names.
- -forceExistingResidueNames¶
Force WRITEDICT to use the given residues, even if they do not match the deposited residues from the RCSB. Also aliased to -force
This flag used to be called -nolookup (which still works) but this was found to be confusing in conjunction with the -lookup flag which does attempts to find the correct residue when there is no match.
- -writeFullDict¶
If true then all refinement dictionaries are created for all residues. If false only residues that are unknown or are in the set of replaceable residues or covalently bound to a ligand are output. Setting this to true can confuse REFMAC5.
{default=false]
- -verbose¶
Output copious information of how writedict operates.
Example Commands¶
The basic usage of writedict is to take an input structure and generate the corresponding .pdb and .cif files for input into COOT or REFMAC5.
> writedict -in 1nhu.ism -out 1nhu_out
This creates the files 1nhu_out.cif and 1nhu_out.pdb. The pdb is always created since the atom names may be remapped from the input. Since the input contains no residue information, the ligand is placed in residue “UNL”.
To add your own residue names for unmapped residues:
> writedict -in 1nhu.ism -out 1nhu_out -residues 153
Note that if the given residue name (153) does not map to the known residue 153 the next residue name in the unmapped residue list will be used. Since none are specified it will go to the standard defaults: “UNL,UN1,UN2...”
Note
When doing whole complexes, renaming residues gets quite complicated since you may not know which residues will map before you run WRITEDICT.
> writedict -in 1nhu.ism -out 1nhu_out -residues LIG -nolookup
Force the residue to be named LIG. (It will also indicate an invalid dictionary)
Warning: LIG 1: Known Residue, but does not map to previous residue: LIG from: RCSB
Warning: LIG 1: previous smiles c1ccc2c(c1)CC3=C(NN=C23)c4ccncc4
Warning: LIG 1: current smiles c1ccc(cc1)CC(C(=O)[O-])N(Cc2cccc(c2)C(F)(F)F)C(=O)c3ccc(cc3Cl)Cl
Warning: LIG: Keeping residue name (dictionary may be invalid)
Warning: LIG: Adding new residue dictionary
Warning: LIG: already in dictionary from: RCSB but with different structure.
Warning: LIG: c1ccc2c(c1)CC3=C(NN=C23)c4ccncc4
Warning: LIG: c1ccc(cc1)CC(C(=O)[O-])N(Cc2cccc(c2)C(F)(F)F)C(=O)c3ccc(cc3Cl)Cl
Warning: LIG: Overwriting existing residue (dictionary may be invalid)
To ensure planarAniline constraints consistent with the MMFF94s forcefield, use the -planarAniline flag.
> writedict -in 1nhu.ism -out 1nhu_out -planarAniline
ALIGNGRID Usage¶
Command Line Interface¶
A description of the command line interface can be obtained by executing ALIGNGRID with the --help option.
> aligngrid --help
will generate the following output:
Help functions:
aligngrid --help simple : Get a list of simple parameters
aligngrid --help all : Get a complete list of parameters
aligngrid --help <parameter> : Get detailed help on a parameter
aligngrid --help html : Create an html help file for this program
Required Parameters¶
- -grid¶
Input density grid. This is the grid that is to be rotated and translated into the new reference frame.
- -out¶
The output grid in any writable format. These include .ccp4, .map and .grd
- -protein¶
The query protein that corresponds to the input grid.
- -target¶
The target protein that the grid and query protein will be aligned toward.
Advanced Options¶
- -outprot¶
An optional file to output the query protein after it is translated and rotated into the new reference frame.
- -padding¶
When writing out a .grd file, this indicates the added extents (in Ångströms around the protein when outputting the density.
- -scale¶
The final map spacing in Ångströms. (The default is the minimum spacing in X, Y or Z)
- -verbose¶
Triggers copious logging output.
MTZ File Parameters¶
- -autoMTZ¶
Automatically try to open the mtz file using the DELWT and FDELWT columns from REFMAC5 mtz files.
[default=true]
- -Fc <columnname>¶
Column to use for Fc. Note, to load arbitrary columns, you can use the *-Fc** and -Phic columns and the Fc maptype.*
[default=FC]
- -Fdelwt <columnname>¶
Column to use for Fdelwt or difference map amplitudes
[default=F]
- -Fobs <columnname>¶
Column to use for FObs.
[default=F]
- -Fwt <columnname>¶
Column to use for Fwt or regular map amplitudes.
[default=FWT]
- -Phic <columnname>¶
Column to use for Phic.
[default=PHIC]
- -Phidelwt <columnname>¶
Column to use for Phdelwt or difference map phases.
- -Phiwt <columnname>¶
Column to use for Phwt or regular map phases.
- -mtype¶
- The map type to use for fitting. Fo-Fc, Fc, 2Fo-Fc, 3Fo-Fc, Fwt,
- Fdelwt...
[default=2Fo-Fc]
Example Commands¶
> aligngrid -grid input.ccp4 -query protein1.pdb \
-target protein2.pdb -out output.ccp4
Take the input grid in ccp4 format and protein. Align protein1 to protein2 and rotate and translate the input grid using the same alignment outputting the result to output.ccp4.
ROTFIT Usage¶
ROTFIT is a side-chain fitting tool that quickly fits sidechains to the surrounding density. Currently only a MTZ file can be used for density Side chains can be fit using several techniques:
- Using the most probably rotamers and ranking based on real space correlation (RSCC) to the density.
- pep-flipping the rotamer and ranking on RSCC.
- Full MMFF/Shape fitting in to the density.
By default ROTFIT searches the rotamers and pep-flips.
For ROTFIT to apply a changed residue, their has to have an increase in RSCC and be above a minium value (currently 0.4). Additionally, the RSCC must be greater by a minimum amount (0.05).
Command Line Interface¶
A description of the command line interface can be obtained by executing ROTFIT with the --help option.
> rotfit --help
will generate the following output:
Help functions:
rotfit --help simple : Get a list of simple parameters
rotfit --help all : Get a complete list of parameters
rotfit --help defaults : List the defaults for all parameters
rotfit --help <parameter> : Get detailed help on a parameter
rotfit --help html : Create an html help file for this program
rotfit --help versions : List the toolkits and versions used in the application
- -in <proteinfile>¶
Input protein structure. The side chains of this protein will be optimized.
- -map <mtz file>¶
The input MTZ file to fit side chains against.
- -out <protein>¶
The protein structure with optimized side chains will be outputted to this file.
- -param <parameter filename>¶
A parameter file is a text file that lists parameter settings to be used during a run. If a parameter is specified both on the command line and in the parameter file, the value specified on the command line is used.
The format of the parameter file is as follows:
- One parameter per line.
- For non-list parameters one key-value pair per line.
- For list parameters a key followed by all the values.
- Boolean parameters must be listed as a key followed by true or false.
- The parameter file may not contain the -param parameter.
- Lines begining with # are considered comments.
- -verbose <true or false>¶
Ouptuts copious extra information.
[ Default : false ]
Advanced Parameters¶
- -fixup_rotamers <true or false>¶
Search the most probable rotamers and choose the one that fits best to the surrounding density.
[ Default : true ]
- -fixup_pepflips <true or false>¶
Search the pep-flips and pick the one that fits best to the surrounding density.
[ Default : false ]
- -fixup_shapefit_residues <true or false>¶
Use MMFF/Shape to fit the residue into the surrounding density. Normally it is best to use -fixup_rotamers in conjunction with this flag.
[ Default : false ]
- -minimum_rscc <number>¶
For a give residue if the best tested RSCC of any conformation is below this value the residue will not be modified. This prevents optimization from happenening when the is no density anywhere near the side chain.
[ Default : 0.4 ]
- -minimum_rscc_increase <number>¶
For a given residue if the best tested RSCC of any conformation is not this much better than the starting conformation, then the residue will not be modified.
[ Default : 0.05 ]
MTZ File Parameters¶
- -autoMTZ <true or false>¶
Automatically try to open the mtz file using the DELWT and FDELWT columns from REFMAC5 mtz files.
[default=true]
- -Fc <columnname>¶
Column to use for Fc. Note, to load arbitrary columns, you can use the *-Fc** and -Phic columns and the Fc maptype.*
[default=FC]
- -Fdelwt <columnname>¶
Column to use for Fdelwt or difference map amplitudes
[default=F]
- -Fobs <columnname>¶
Column to use for FObs.
[default=F]
- -Fwt <columnname>¶
Column to use for Fwt or regular map amplitudes.
[default=FWT]
- -Phic <columnname>¶
Column to use for Phic.
[default=PHIC]
- -Phidelwt <columnname>¶
Column to use for Phdelwt or difference map phases.
- -Phiwt <columnname>¶
Column to use for Phwt or regular map phases.
- -mtype <setting>¶
- The map type to use for fitting. Fo-Fc, Fc, 2Fo-Fc, 3Fo-Fc, Fwt,
- Fdelwt...
[default=2Fo-Fc]
RSCC Usage¶
RSCC is a real-space fit calculation tool both real-space correlation coefficient (rscc) and real-space R-factor (rsr). By default this tool calculates rscc. RSCC was designed to be a standalone tool using the same code that is used for ligand fitting by AFITT.
- -in <molecule file>¶
Input molecule(s) to be scored against the density
- -mtz <mtz file>¶
Input MTZ file to be used for calculations. Currently only MTZ format is supported.
Advanced Parameters¶
- -clamp_at_level¶
Set an electron density contour sigma level (positive) to be used for calculating rsr values. The default value is 0.0.
- -flatten¶
Flatten B-factors (20.0) of the input molecule. This flag is to be used with the –scatter flag.
- -rsr¶
Use rsr instead of rscc.
- -scatter¶
Calculate Fc (calculated) density from the input molecule.
- -verbose¶
This triggers copious logging of output to the screen.