Choosing Data: Molecule/Maps Page¶
The molecules/maps page is where data is loaded into AFITT. The basic overview of this page is shown in Figure: AFITT Molecules Page
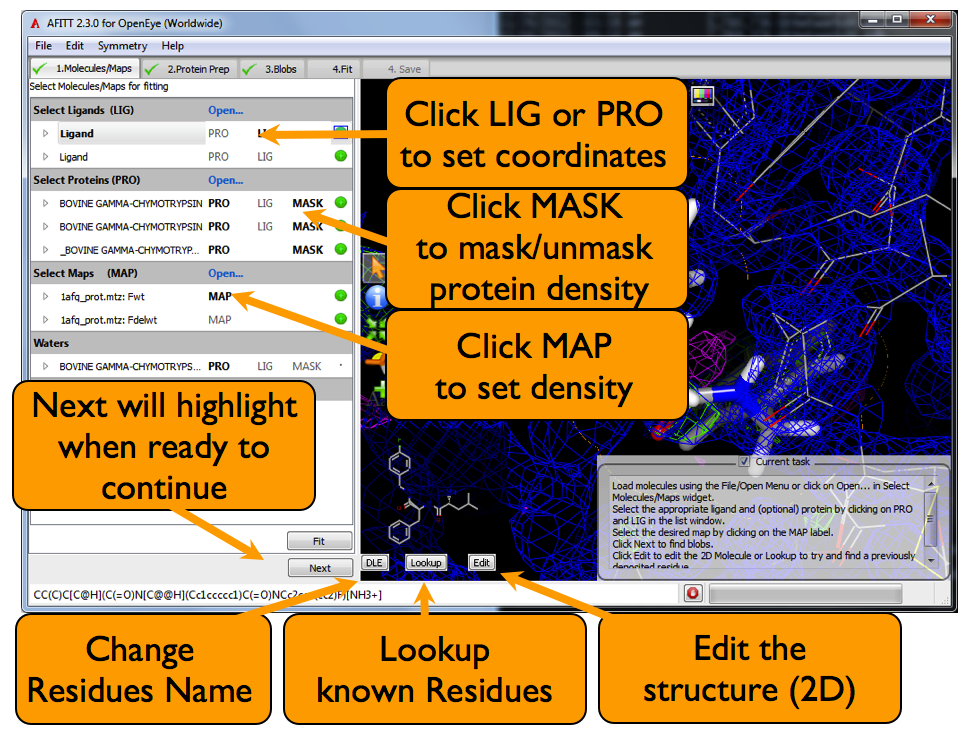
AFITT Molecules Page
AFITT automatically seperates ligand-like molecules from protein-like molecules and attempts a reasonable first guess as to whether they are ligands or proteins.
Selecting Ligands Or Proteins¶
Clicking on the LIG text in the list window will select a ligand to be fit into density. Clicking on the PRO text will select the proteins used for both masking density and for the purposes of refinement.
Note
Multiply ligands can be fit simultaneously, simply shift-click to select a range of ligands and click LIG or hold shift and individually click LIG on the desired ligands.
Note
If the LIG has a line through it, then the MMFF force field does not support the atom types of the ligand and cannot be fitted with the current version of AFITT. An example is shown in figure Ligand that cannot be fit.

Indication that a ligand cannot be fit by AFITT
Selecting the ligand by clicking on the name in the list window will make it active. Active molecules can be centered by clicking on the center button at the top of the 3D window. Active molecles are highlighted by a surrounding yellow halo.
Note
2D molecules loaded from smiles strings or other connection table formats will not have a halo in the 3D window since they will not have a 3D representation. Their depictions, however, will show up in the lower left of the screen.
The currently selected ligand is shown in the lower left corner of the 3D window.
Loading Maps¶
Maps are loaded either through the File/Open... menu, the File/Open Maps... menu or by clicking “Load” in the list window MAP section as seen in Figure: AFITT Molecules Page.
AFITT handles most common maps types including:
- .mtz maps
- .ccp4 maps
- various other reflection formats: .hkl, .phs, .fobs, .cv or .cif
- OpenEye ascii grids .grd
MTZ Maps¶
By default, when opening a CCP4 .mtz map, the default columns and maps are chosen, either FWT and DELFWT maps or 2Fo-Fc and Fo-Fc maps.
To have more control over which maps are created, use the File menu option Open MTZ.... This opens a dialog (as seen in figure Figure AFITT MTZ Open Dialog) that allows selection of both the MTZ columns and the desired output maps.
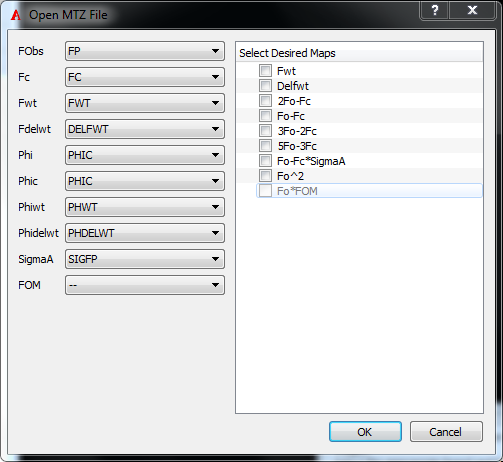
AFITT MTZ Open Dialog
Simply select the appropriate columns and map types to generate and click “OK”.
Looking Up Residues¶
Residue information is often lost, especially when loading from file formats that do not have connection tables. Refinement programs such as REFMAC5 are very finicky when supplied with residue and atom labels for refinement dictionaries that are already known or come pre-loaded into the refinement program.
For instance, if your atom names do not exactly match up with the atom names in a REFMAC5 refinement dictionary, you may fail to produce any refinement.
Clicking on the “Lookup” button searches more than 5,000 previously refined ligand structures. If AFITT finds a compatible match, a dialog is shown that allows assignment of the residue name. If a residue match is found, the atom names of the ligand are modified to match the known refinement dictionary guaranteeing a successful refinement.
An example of a succesful match is seen in Figure Lookup Known Residues. In this case, two compatible refinment dictionaries are found, AZM and MZM which are very close analogs. MZM is deemed to be compatible becuase it is a super-structure of the initial ligand. To select the desired residue, click on the checkbox next to the depiction. Otherwise hit cancel to not set a residue name.
Note
This doesn’t mean that MZM is the correct refinement dictionary however. It just guarantees that refmac will not complain and throw an error during a refinement.
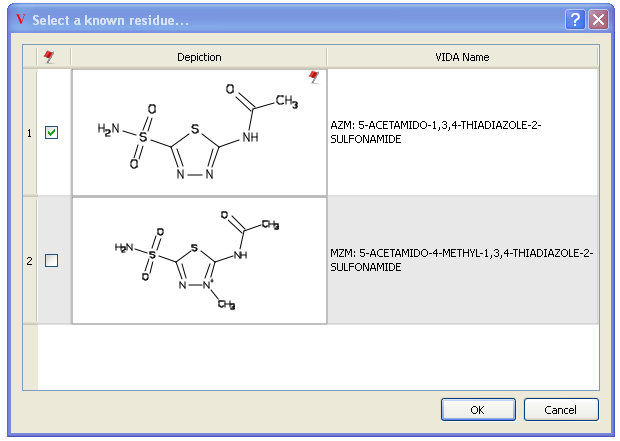
AFITT Lookup up known residues
Note
AFITT names unfound residues as “UNL” which is the PDB code for Unknown Ligand. Of course, one should never deposit a structure with this residue name.
A good example of a refinement dictionary lookup failure is seen in figure Figure AFITT Refinemnt Dictionary Failure
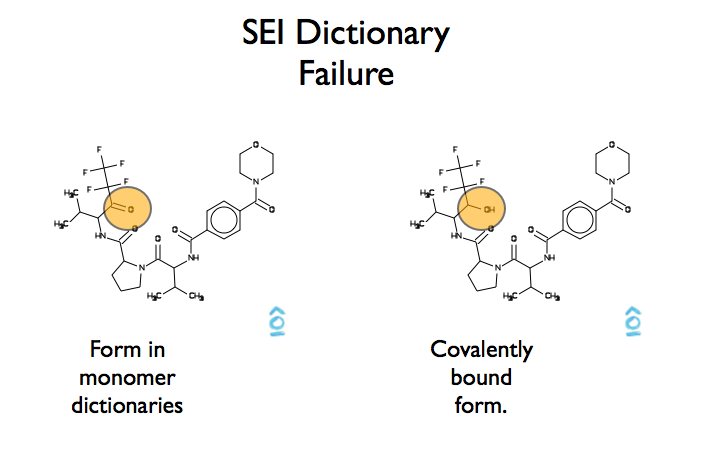
AFITT Refinement Dictionary Failure
The structure of SEI as stored by REFMAC5 is the non-covalent ketone form. However because SEI forms a covalent bond in pdb id 1B0F, AFITT is unable to locate a compatible residue dictionary simply because the covalently bound form has an extra hydrogen atom not found in REFMAC5’s refinement dictionary. In cases like these, a new dictionary has to be generated.
Editing a Ligand¶
Clicking on the edit button opens up AFITT’s molecule sketcher where the connection table of the initial ligand can be edited. When the editing is completed, a new molecule is added to AFITT; the original ligand is not changed.
A basic overview of the 2D sketcher can be seen in Figure Basic overview of 2D Sketcher
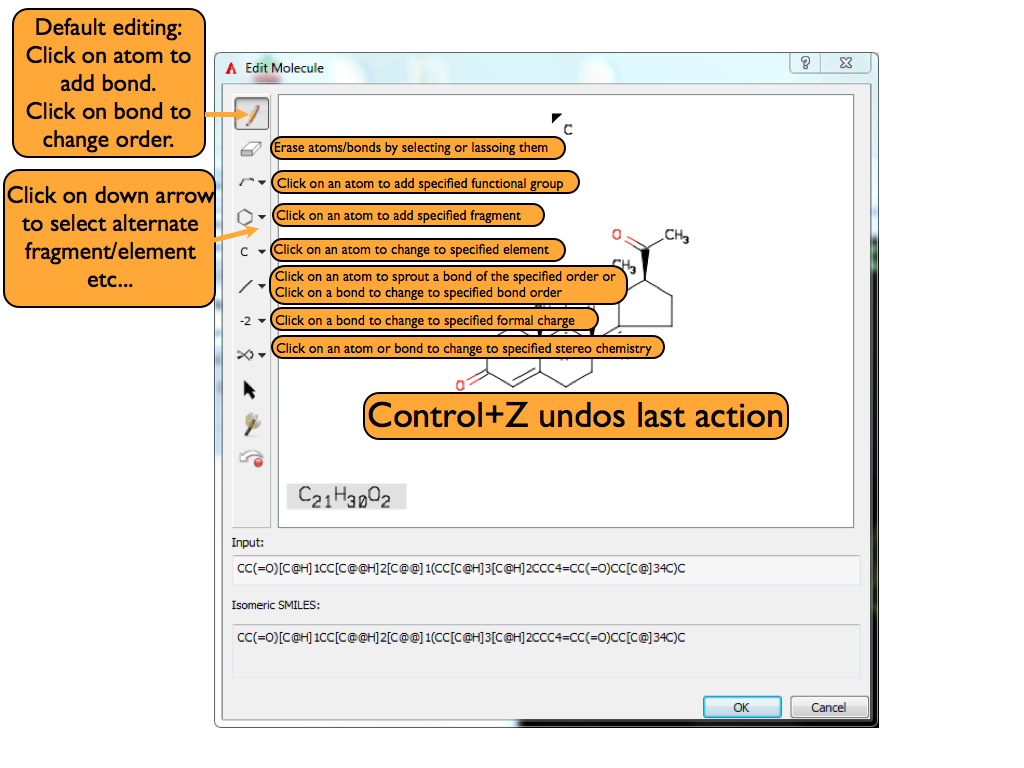
Basic overview of 2D sketcher
Whenever a ligand is edited, the original ligand is retained and a new molecule is created. The new molecule does not retain any residue information from the original molecule.