Visualizing Electron Density¶
Problem¶
You want to visualize how a ligand fits into an electron density grid in a 2D molecule diagram (see Figure 1). The electron density grid with various contour levels and the 3D molecule is shown in Table 1.
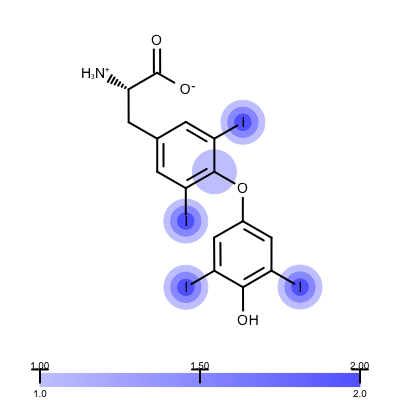
Figure 1. Example of visualizing ligand electron density fit (PDB: 1ETS)
| contour 1.0 | contour 1.5 | contour 2.0 |
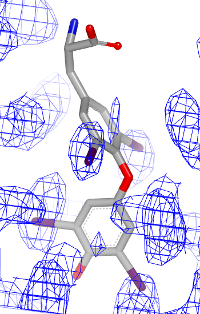
|
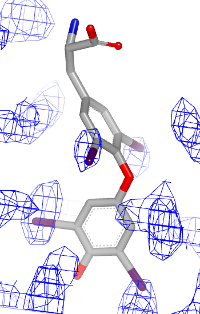
|
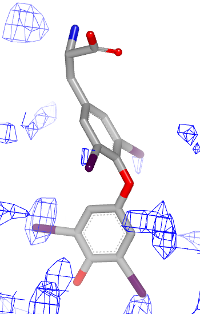
|
Ingredients¶
|

Download¶
Solution¶
The set_electron_density_contour_overlap function tags an atom if its coordinates inside the grid at the given contour level.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 | def set_electron_density_contour_overlap(mol, edgrid, contour, contour_tag):
"""
Sets whether atoms are inside the electron density grid at the given
contour level.
:type mol: oechem.OEMolBase
:type edgrid: oegrid.OESkewGrid
:type contour: float
:type: contour_tag: int
"""
center = oechem.OEFloatArray(3)
extents = oechem.OEFloatArray(3)
oechem.OEGetCenterAndExtents(mol, center, extents)
subgrid = oegrid.OEScalarGrid()
# expand the grid a bit for proper overlaps
extents[0] += 2.5
extents[1] += 2.5
extents[2] += 2.5
oegrid.OEMakeRegularSubGrid(subgrid, edgrid, center, extents, 0.5, edgrid.GetReentrant() >= 7)
for atom in mol.GetAtoms(oechem.OEIsHeavy()):
xyz = mol.GetCoords(atom)
val = subgrid.GetValue(xyz[0], xyz[1], xyz[2])
if val > contour:
atom.SetData(contour_tag, val)
|
The depict_erlectron_density_fit function shows how to project the electron density fit with various contour levels into a 2D molecule diagram. First the image is divided into two image frames since both a molecule and a color gradient will be depicted. Then the set_electron_density_contour_overlap function is called that calculates whether the atoms of the molecule are inside the electron density grid at various contour levels (see lines 21-23). The molecule is then prepared for depiction generating its 2D coordinates by calling the OEPrepareDepictionFrom3D function (lines 27-32). After constructing a color gradient that will assign colors to various contour levels, the function loops over the atoms and draws a circle around them if they are embedded into the electron density grid (> 0.2) with a color and radius that corresponds to the given contour level (lines 42-51). Finally, the molecule along with the color gradient is rendered to the image (lines 55-62). You can see the result in Figure 1.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 | def depict_electron_density_fit(image, mol, edgrid, opts):
"""
Depicts the molecule with electron density fit.
:type image: oedepict.OEImageBase
:type mol: oechem.OEMolBase
:type edgrid: oegrid.OESkewGrid
:type opts: oedepict.OE2DMolDisplayOptions
"""
# generate image frames
width, height = image.GetWidth(), image.GetHeight()
mframe = oedepict.OEImageFrame(image, width, height * 0.90,
oedepict.OE2DPoint(0.0, 0.0))
lframe = oedepict.OEImageFrame(image, width, height * 0.10,
oedepict.OE2DPoint(0.0, height * 0.90))
# calculate fit to electron density at various contours
contours = [1.0, 1.5, 2.0]
for contour in contours:
set_electron_density_contour_overlap(mol, edgrid, contour, get_contour_tag(contour))
# prepare molecule for depiction
width, height = mframe.GetWidth(), mframe.GetHeight()
opts.SetDimensions(width, height, oedepict.OEScale_AutoScale)
oegrapheme.OEPrepareDepictionFrom3D(mol)
opts.SetScale(oedepict.OEGetMoleculeScale(mol, opts) * 0.95)
disp = oedepict.OE2DMolDisplay(mol, opts)
# create color gradient
colorg = oechem.OELinearColorGradient()
colorg.AddStop(oechem.OEColorStop(min(contours), oechem.OEColor(190, 190, 255))) # light blue
colorg.AddStop(oechem.OEColorStop(max(contours), oechem.OEColor(80, 80, 255))) # medium blue
# visualize electron density fit
layer = disp.GetLayer(oedepict.OELayerPosition_Below)
for contour in contours:
contour_tag = get_contour_tag(contour)
radius = get_contour_radius(contour, contours, disp)
color = colorg.GetColorAt(contour)
pen = oedepict.OEPen(color, color, oedepict.OEFill_On, 1.0)
for atom in mol.GetAtoms():
if atom.HasData(contour_tag):
adisp = disp.GetAtomDisplay(atom)
layer.DrawCircle(adisp.GetCoords(), radius, pen)
# render molecule
oedepict.OERenderMolecule(mframe, disp)
# draw color gradient
copts = oegrapheme.OEColorGradientDisplayOptions()
copts.SetColorStopPrecision(1)
copts.AddMarkedValues(contours)
oegrapheme.OEDrawColorGradient(lframe, colorg, copts)
|
Usage¶
Usage
elecdensity2img.py and supporting data 1eta_sigma.mtz and 1eta_ligand.pdb
prompt > python3 elecdensity2img.py -grid 1eta.mtz -ligand 1eta_ligand.sdf -out 1eta.png
Command Line Parameters¶
Simple parameter list
-height : Height of output image
-width : Width of output image
molecule display options :
-titleloc : Location of the molecule title
input/output options
-ligand : Input filename of the ligand
-grid : Input filename of electron density map (MTZ)
-out : Output filename of the generated image
Discussion¶
Visualizing electron density helps to evaluate the quality of protein-ligand structures. The Iridium database divides the protein-ligand structures into three categories:
- Iridium-NT (not trustworthy)
- Iridium-MT (moderately trustworthy)
- Iridium-HT (highly trustworthy)
Not surprisingly the 1ETS complex (Figure 1 is considered “not trustworthy” by the Iridium database. The Table 2 shows examples from the MT and HT categories of the Iridium database
| 1COY - Iridium MT | 1D3H - Iridium HT |
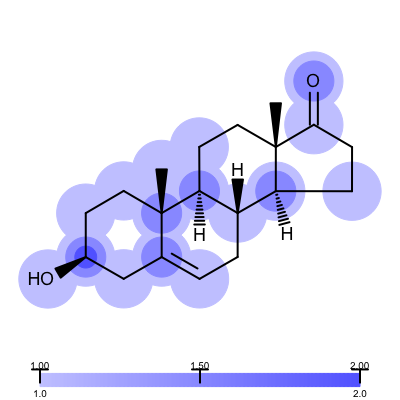
|
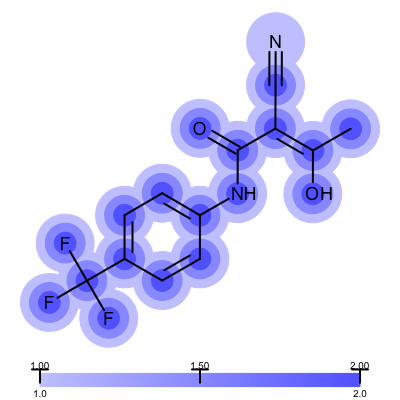
|
The visualization also helps to compare the “deposited” starting models of the Iridium database with the models that are refined by AFITT. See example in Table 3.
| deposited starting model | model after AFITT refinement |
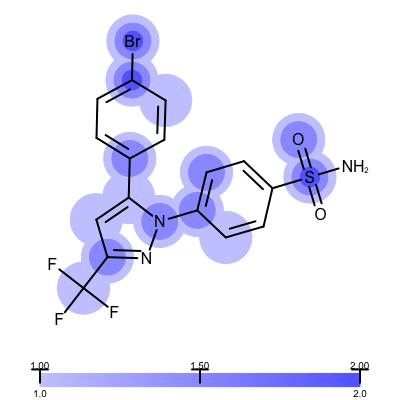
|
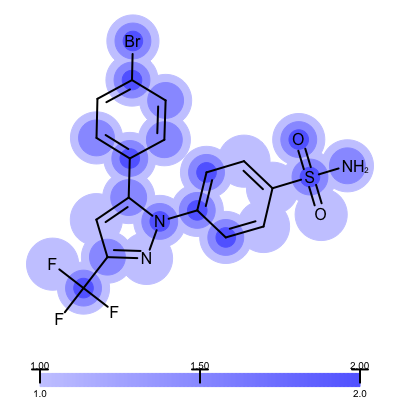
|
Command Line Parameters¶
Simple parameter list
input/output options
-complex : Input filename of the protein-ligand complex
-out : Output filename of the generated image
residue options
-ignore-isolated : Removes isolated atoms prior to depiction
-ignore-water : Removes water molecule prior to depiction
-max-residues : Controls the maximum number of residues depicted in each
line
See also in OEChem TK manual¶
API
- OEColorStop class
- OELinearColorGradient class
- OEMakeRegularSubGrid function
- OEScalarGrid class
Theory
- OEGrid Theory chapter
See also in OEDepict TK manual¶
Theory
- Molecule Depiction chapter
API
- OE2DMolDisplay class
- OE2DMolDisplayOptions class
- OEImage class
- OEImageFrame class
- OERenderMolecule function
See also in GraphemeTM TK manual¶
API
- OEPrepareDepictionFrom3D function
- OEColorGradientDisplayOptions class
- OEDrawColorGradient function