Enumerating Fragment Combinations¶
Problem¶
You want to enumerate all the adjacent fragment combinations returned by the fragmentation methods of OEMedChem TK.
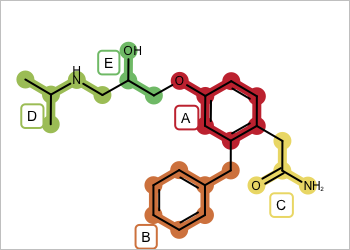
Figure 1: Example molecule with fragment highlighting
For example in Figure 1 you want to return fragment combinations: A+B, B+C, ..., A+B+C, ..., A+B+C+D, ... etc. But you do not want disconnected fragment combinations such as D.A+B. See examples in Table 1.
| valid - all fragments adjacent | invalid - disconnected fragments |
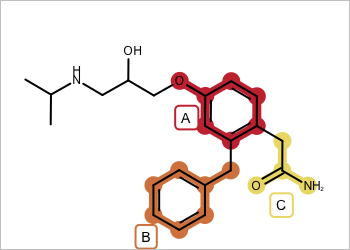
|
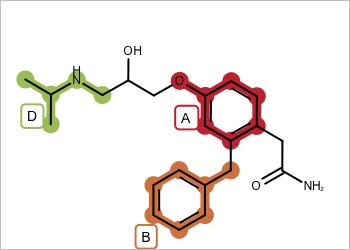
|
Ingredients¶
|

Solution¶
The OEMedChem TK currently provides three ways to partition a molecule into fragments (see examples in Table 2):
- The OEGetRingChainFragments function fragments a molecule into ring and chain components.
- The OEGetRingLinkerSideChainFragments function fragments a molecule into ring, linker and side-chain components as defined in [Bemis-1996] .
- The OEGetFuncGroupFragments function fragments a molecule into ring and functional group components.
| OEGetFuncGroupFragments | OEGetRingChainFragments | OEGetRingLinkerSideChainFragments |
 |
 |
 |
The GetFragmentCombinations function takes a molecule and the list of OEAtomBondSet objects. This list is generated by using one of the fragmentation methods shown in Table 2. Each OEAtomBondSet object stores the atoms and the bonds of a fragment generated for a given molecule. These parameters are passed to the GetFragmentAtomBondSetCombinations function that returns the list of adjacent fragment combinations. Each fragment combination is then passed to the OESubsetMol function using an OEIsAtomMember and an OEIsBondMember predicate. The subset molecule generated by the OESubsetMol function is then appended to a returned molecule list.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | def GetFragmentCombinations(mol, fraglist):
fragments = []
fragcombs = GetFragmentAtomBondSetCombinations(mol, fraglist)
for f in fragcombs:
fragatompred = oechem.OEIsAtomMember(f.GetAtoms())
fragbondpred = oechem.OEIsBondMember(f.GetBonds())
fragment = oechem.OEGraphMol()
adjustHCount = True
oechem.OESubsetMol(fragment, mol, fragatompred, fragbondpred, adjustHCount)
fragments.append(fragment)
return fragments
|
The GetFragmentAtomBondSetCombinations function below generates all fragment combinations in the range of [2 - (nrfrags-1)] using the itertools.combinations() function. For example, if there are five fragments generated for a molecule, then the following number of fragment combinations are enumerated:
\(\dbinom{5}{2} + \dbinom{5}{3} + \dbinom{5}{4} = \frac{5!}{2!3!} + \frac{5!}{3!2!} + \frac{5!}{4!1!} = 10 + 10 + 5 = 25\)
However, only those fragment combinations that are adjacent will be kept i.e. no disconnected molecule fragment is generated (see Table 1). If the fragments in a given combination are all adjacent i.e. the IsAdjacentAtomBondSetCombination function returns true, then the atoms and the bonds of these fragments are combined by calling the CombineAndConnectAtomBondSets function and the combined fragment is added to the list returned by the GetFragmentAtomBondSetCombinations function.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 | def GetFragmentAtomBondSetCombinations(mol, fraglist):
fragcombs = []
nrfrags = len(fraglist)
for n in range(2, nrfrags):
for fragcomb in combinations(fraglist, n):
if IsAdjacentAtomBondSetCombination(fragcomb):
frag = CombineAndConnectAtomBondSets(fragcomb)
fragcombs.append(frag)
return fragcombs
|
A fragment combination is considered adjacent only if all of the fragments are connected to each other in the combination. In order to determine this the IsAdjacentAtomBondSetCombination function performs a depth-first search algorithm using the TraverseFragments function.
The nrparts variable keeps track of the number of connected components found in the fragment list. If the fragments are all connected than nrparts will be one.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | def IsAdjacentAtomBondSetCombination(fraglist):
parts = [0] * len(fraglist)
nrparts = 0
for idx, frag in enumerate(fraglist):
if parts[idx] != 0:
continue
nrparts += 1
parts[idx] = nrparts
TraverseFragments(frag, fraglist, parts, nrparts)
return (nrparts == 1)
|
The TraverseFragments is a recursive function that visits fragments that are connected to each other. The parts list is used not only to keep track of whether a fragment has been visited before, but also to which connected fragment component it belongs.
1 2 3 4 5 6 7 8 9 10 11 | def TraverseFragments(actfrag, fraglist, parts, nrparts):
for idx, frag in enumerate(fraglist):
if parts[idx] != 0:
continue
if not IsAdjacentAtomBondSets(actfrag, frag):
continue
parts[idx] = nrparts
TraverseFragments(frag, fraglist, parts, nrparts)
|
The IsAdjacentAtomBondSets function is used to determine whether two fragments are connected. Two fragments are considered adjacent if an atom from each is connected by a bond.
1 2 3 4 5 6 7 | def IsAdjacentAtomBondSets(fragA, fragB):
for atomA in fragA.GetAtoms():
for atomB in fragB.GetAtoms():
if atomA.GetBond(atomB) is not None:
return True
return False
|
If the IsAdjacentAtomBondSetCombination function returns true i.e the fragments are all connected, then the CombineAndConnectAtomBondSets function can be used to combine the adjacent fragments together by merging their atoms and bonds and adding those bonds that connect them.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | def CombineAndConnectAtomBondSets(fraglist):
# combine atom and bond sets
combined = oechem.OEAtomBondSet()
for frag in fraglist:
for atom in frag.GetAtoms():
combined.AddAtom(atom)
for bond in frag.GetBonds():
combined.AddBond(bond)
# add connecting bonds
for atomA in combined.GetAtoms():
for atomB in combined.GetAtoms():
if atomA.GetIdx() < atomB.GetIdx():
continue
bond = atomA.GetBond(atomB)
if bond is None:
continue
if combined.HasBond(bond):
continue
combined.AddBond(bond)
return combined
|
Download code
The following example shows the output fragment combinations of the molecule depicted in Figure 1.
Usage:
prompt > python3 enumfrags.py -in .ism -out .ism -fragtype funcgroup
CC(C)NCC(COc1ccc(c(c1)Cc2ccccc2)CC(=O)N)O
5 number of fragments generated
11 number of fragment combinations generated
c1ccc(cc1)Cc2cccc(c2)O
c1cc(ccc1CC(=O)N)O
c1ccc(cc1)OCCO
CC(C)NCC(C)O
c1ccc(cc1)Cc2cc(ccc2CC(=O)N)O
c1ccc(cc1)Cc2cccc(c2)OCCO
c1cc(ccc1CC(=O)N)OCCO
CC(C)NCC(COc1ccccc1)O
c1ccc(cc1)Cc2cc(ccc2CC(=O)N)OCCO
CC(C)NCC(COc1cccc(c1)Cc2ccccc2)O
CC(C)NCC(COc1ccc(cc1)CC(=O)N)O
See also in Python documentation¶
See also in OEChem TK manual¶
API
- OEAtomBondSet class
- OEIsAtomMember predicate
- OEIsBondMember predicate
- OESubsetMol function
See also in OEMedChem TK manual¶
Theory
- Molecule Fragmentation chapter
API
- OEGetFuncGroupFragments function
- OEGetRingChainFragments function
- OEGetRingLinkerSideChainFragments function