Fit Ligand: Fit Page¶
Entering the Fit page for the first time starts the automatic fitting process against the previously selected regions of density. Once a fit is completed, the best fit conformation for each selected blob is shown and selected for refinement or saving.
The basic fitting page is shown in figure Fitting Page.
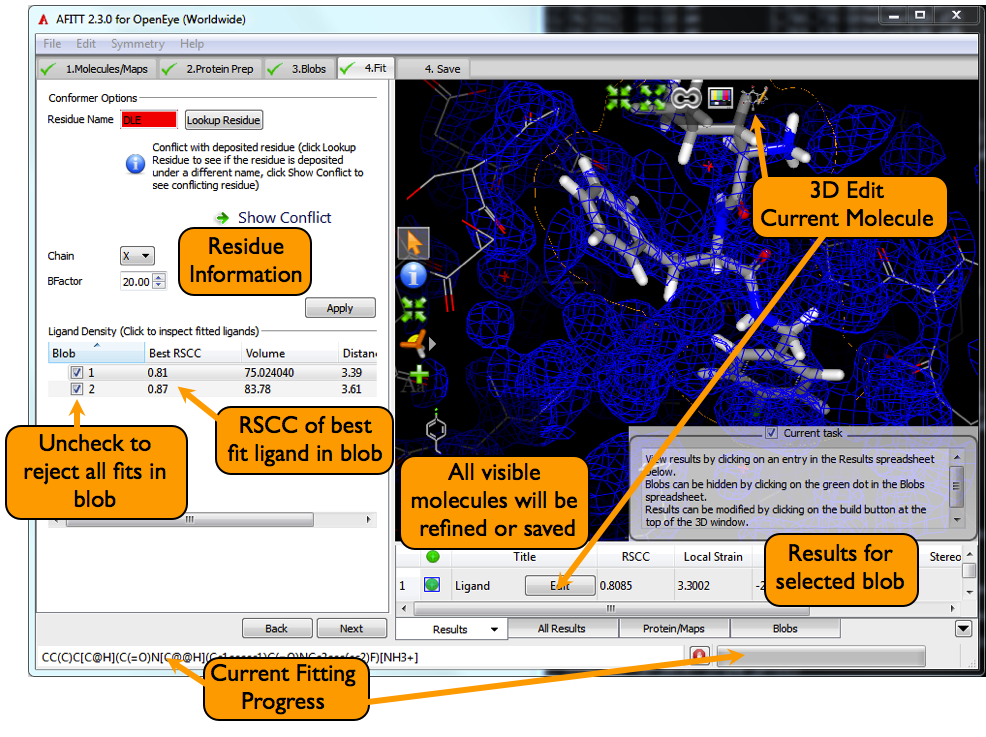
AFITT Fit Ligand Page
Looking at Results¶
Specific details about the results are shown in the Results tab in the spreadsheet. To investigate the results, click on the desired blob from the Blobs Fit Against list seen in Figure Fitting Page. The results spreadsheet will be populated with the ligands fitted against the selected blob.
The basic overview of the spreadsheet can be seen in Figure Results Spreadsheet.

Ligand fitting results For RSCC, higher is better, for PLP and Chemscore, lower scores are better.
By default, the results are sorted against RSCC (real space correlation coefficient), which is a standard measure of how the ligand fits against the density. Additionaly, poses are scored with PLP and Chemscore. The strain energies calculated with AFITT are always compared against the lowest energy conformation in the conformational ensemble as determined by the fitting process. This is equivalent to the technique used by Perola and Charifson. [Perola-2004].
AFITT uses two protein environment (docking) scores to help distinguish between conformations and placements of ligands (and fragments) which have nearly identical real-space correlation coefficient (RSCC) values, (this can happen easily when fitting symmetric molecules). The two scoring functions (PLP and Chemscore) used by AFITT were chosen because they were designed using very different strategies to solve different problems in molecular docking. Chemscore [Eldridge-1997] was designed to estimate free energy of binding and contains a hydrogen bonding, a metal-ligand, a lipophilic (van der Waals interaction) and a rotatable bond (entropy) term. The coefficients of these terms were optimized against a protein-ligand data set to predict measured binding affinities. Note that this function was not designed or optimized to predict the correct binding mode but rather protein-ligand binding affinity. The piece-wise linear potential (PLP) [Verkhivker-2000] scoring function was derived by generating a potential for a set of heavy atom pairs observed in the Protein Data Bank (PDB) at the time of publication (2000). The heavy atoms pairs were used to generate steric, hydrogen bond (traditional and sulfur-hydrogen), metal chelating, halogen and repulsive terms. PLP was tested and optimized to predict the geometry of the bound ligand. The advantage of using scoring methods that are in their design orthogonal is that when the predictions agree there is a higher probability that the prediction is correct. Our experience with these scores is that when compared individually with RSCC they select the correct conformation and placement only about 50% of the time. However, when the RSCC values for a set of fits are very similar (differing by less than 0.05), and both scoring functions agree that a particular fit is better there is a high probability that that fit is the correct one. Many ligands are not very symmetric so examples where the RSCC values for different fits are similar are rare. However, when fitting highly symmetric molecules or small, lightly functionalised fragments, RSCC values for different fits can be very similar. In these cases protein environment (docking) scores are critical for successfully identifying the correct fit.
Selecting a ligand will make it active and produce a yellow halo around the molecule. The halo is the extent of the molecular surface and can be used to visually inspect the fit against density.
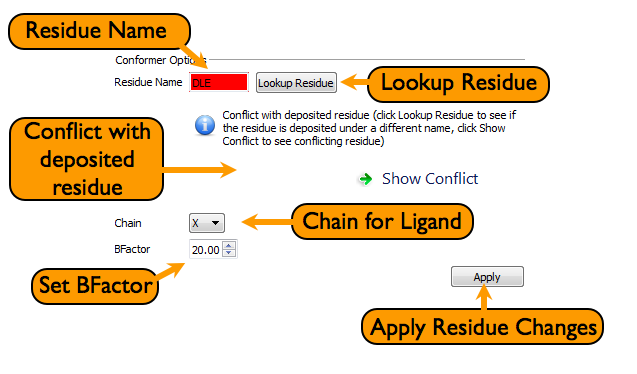
Changing Ligand Residue Information¶
AFITT checks to see if the current residue information conflicts with a known deposited residue. This conflict can be as simple as an incorrect atom name or can be as serious as conflicting connectivity.
Note
When fitting multiple ligands, AFITT does not allow residue names to be altered at this stage.
At this stage, AFITT will not automatically change residue names but does indicate errors as seen in figure Residue Error.
To resolve conflicts, residue information of the fitted ligand can be changed at this point, including:
- residue name
- Chain ID
- B-factor
Clicking the Lookup button will run the residue lookup as described in Looking up Residues. The residue name can also be entered by hand in the entry widget as seen in figure Fitting Page.
If a ligand is found to be incompatible with an existing refinement dictionary from RCSB or REFMAC, a warning is placed near the residue name as seen in figure Residue name error.
The existing residue has a conflict with a known residue
AFITT will not alter the residue name until it generates a refinement dictionary.
Selecting Ligands For Refinement¶
By default, the best fit ligand for each blob is set to be visible and, hence, is selected for refinement or saving. To select an alternate ligand or to have multiple conformations, select the desired blob from the Blobs Fit Against window, and then click on the Results tab in the spreadsheet if it isn’t already selected.
This will present a list of ligands fit to the selected density region as seen in figure Results Spreadsheet.
Note
If multiple ligands are visible for a single blob, they will be combined as alternate conformations with an occupancy of 1/N where N is the number of visible ligands for the blob.
When a ligand is selected in the list window, a molecular surface halo appears surrounding the ligand. This halo is quite useful when judging the fit of the ligand against density. As seen in figure Fitting Halo, the outline of the molecular surface should follow the outline of the density for good fits.
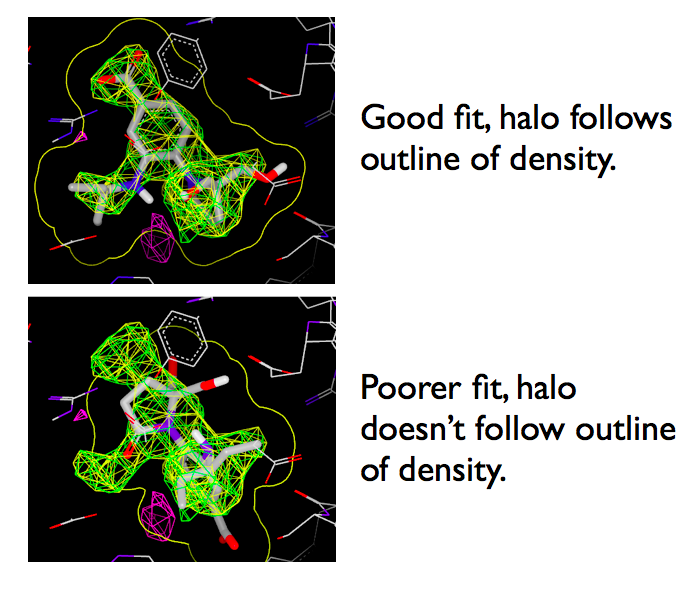
Fitted Halo shows goodness of Fit
Covalent Bonds¶
If any of the ligand’s heavy atoms are significantly close to the protein, a covalent bond is added to the potential covalent bond list. Clicking on a covalent bond in the list will highlight the proposed bond in the 3D window as seen in figure figure Highlighted Covalent Bond<figure_highlighted_bond.
Note
Only one covalent bond and one ligand conformation can be used at a time.
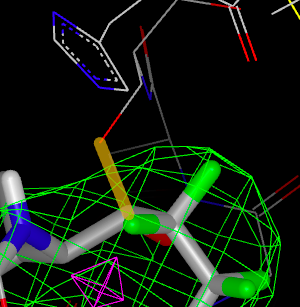
Highlighted Covalent Bond
To select a covalent bond, simply check the associated checkbox. When a covalent bond is selected, the rest of the wizard becomes grayed out because only one ligand may be used for the complex.
Unselecting the checkbox will remove the covalent bond and also undo any ligand editing or minimization leaving the ligand and protein in their original states.
Editing Ligands¶
Fitted ligands can also be edited at this point. Clicking on the 3D Edit Current Molecule button as shown in figure Fitting Page starts AFITT’s integrated molecule builder.
When a ligand is edited, AFITT always edits a copy of the ligand so both original molecule and the edited version will be available for refinement or saving. For more details on molecular editing, please see the section on Molecular Editing.