Protein Preparation¶
Protein preparation is an optional workflow that cleans up residue geometries utilizing their local density. Protein preparation is off by default, it may be enabled by opening AFITT’s preferences and ensuring the Fix Geometry checkbox is checked. See figure Workflow Preferences.
This is useful when analyzing co-crystals which may have slightly different residue geometries.

AFITT Protein Preparation Page
Local residue geometries are fixed using the following methods:
- Rotamer search using the Richardson rotamer libraries.
- Peptide flip searching.
- Full MMFF94s geometry optimization.
The default method is rotamer searching.
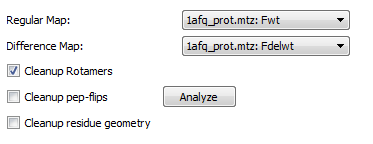
AFITT Protein Preparation Geometry Options
The alternate side-chain positions generated from protein preparation are compared by real space correlation coefficient (RSCC). In cases where there is limited density to define the side-chain position, the orientation that best avoids any negative density is preferred.
Applying New Residue Geometries¶
After the side chain geometries have been cleaned up, the orientations are compared as described above and the best scoring orientation is placed in the residue list. To apply these suggested changes, select the desired residues and click the Apply button.
If a difference map is selected, the difference map is applied to the regular density to help guide the new placement.
Note
Because a difference map contains positive and negative density, you may occasionally see a slightly negative RSCC. This indicates that atoms have been placed near negative density in the omit map.
Inspecting Modified Residues¶
To inspect a particular residue, click on the residue in the list to center on the residue. The new residue geometry is highlighted in orange. To select the new geometry, check the residue in the list.

AFITT Protein Preparation Residue Navigation. The Initial and Final columns indicate the change in goodness of fit (real space calculation based on both regular and difference maps)
Clicking Apply will apply the new geometry to the protein.