Automate Protein Structures Preparation and Selection Process (November 23, 2021)
Protein structures need to be prepared before use in docking or molecular dynamics (MD) calculations. In Orion, Spruce floes simplify the process for you and help you select the best structure to use.
With Spruce floes, you can quickly prepare proteins by providing PDB files or PDB accession codes as input.
You may prepare several related proteins and use Spruce to align all structures to a user-provided reference structure, allowing you to easily compare the interactions.
You may prepare your protein structures with non-OpenEye tools and import those prepared proteins into Orion; Spruce will inform you of any potential problems that still exist.
Why Should I Prepare Protein Structures Prior to Use in Docking or MD?
A lot of protein structures cannot be used in downstream applications as they are, because the information they contain is incomplete:
Hydrogen atoms, heavy atoms in protein side chains, even entire residues can be missing.
The source file may not contain the relevant oligomerization state.
Partial charges may be missing, or incorrect tautomerization states could be present.
Spruce floes can help you resolve those problems (see Spruce Theory).
How Does Spruce Automate the Process for Me?
The Spruce floes offered in Orion take a protein structure as input, either as an input file in PDB or MMCIF format or as a PDB accession code, and generate a design unit or multiple design units as output. A DU contains the protein and ligand (if available), as well as other information required for use in docking or MD applications, such as solvent, packing residues, binding site information, and, if provided, electron density maps (2Fo−Fc and Fo−Fc) for the binding site. Note that the Spruce floes can prepare multiple structures simultaneously and optionally superpose them to a reference structure provided by the user.
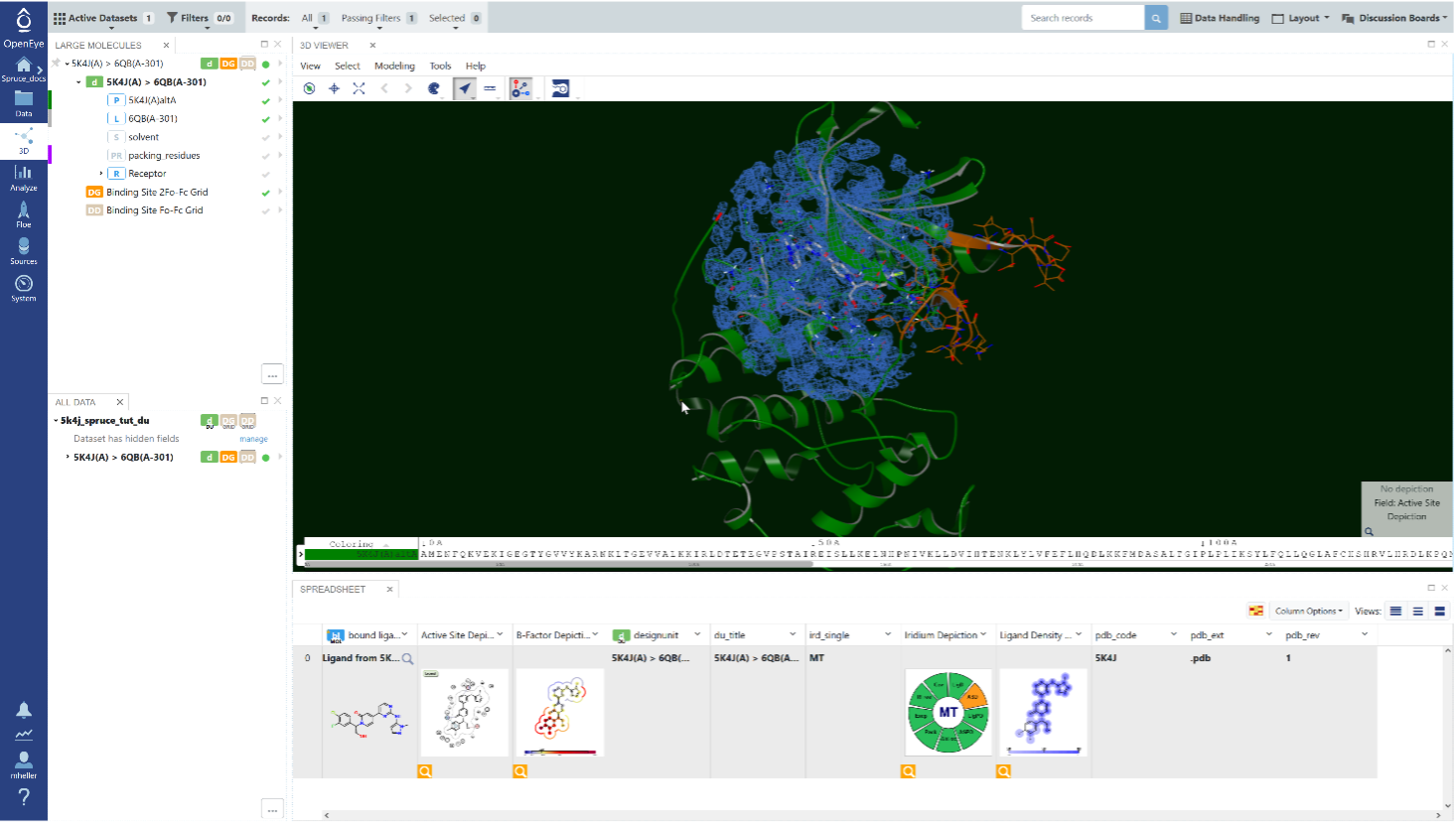
A design unit (DU) prepared with Spruce Floes. Protein residues are shown in green, while residues that were altered by Spruce, or loops that were modeled, are shown in orange. The bound ligand is shown in stick representation, and the 2Fo-Fc grid for the binding site is shown as blue mesh.
How Can I Decide Which Protein Structure to Use for Docking If I Have Multiple Structures?
Spruce not only fixes issues with the protein structure itself but also pays attention to details of critical importance to docking, such as:
How well defined is the electron density for the ligand and the binding site residues?
Are there any excipients close to the binding site that might affect ligand binding?
Are residues from neighbors in the crystal close to the binding site?
Spruce will not only address those questions but also will generate useful depictions, for example, an interaction map of the active site, and the electron density of the ligand. If available, Spruce will use electron density (ED) maps to calculate the Iridium score, which classifies proteins as highly trustworthy (HT), medium trustworthy (MT), or not trustworthy (NT), helping you choose the best structure(s) with confidence for your needs.

The dataset containing four design units (DUs) prepared by Spruce floes for the PDB accession code 4ZJI. The structure contains four chains, with four copies of the ligand bound, hence Spruce created four DUs. The depictions make it easier for the user to pick the appropriate DUs. Note that the upper two DUs have an Iridium score of HT (highly trustworthy), whereas the lower two have an Iridium score of MT (medium trustworthy).
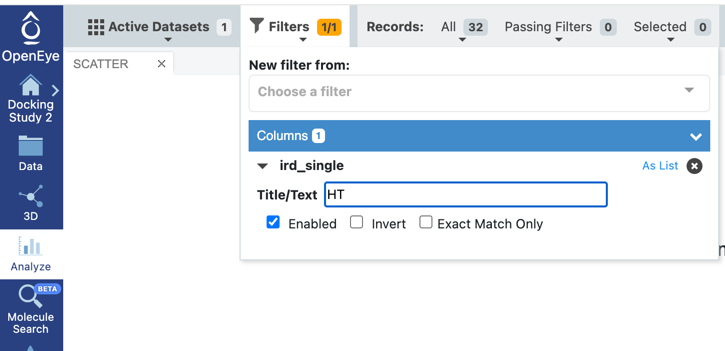
Orion’s filtering functionality allows you to narrow down the number of DUs. For example, you could choose to only display those with an Iridium score of HT (highly trustworthy), making structure selection an easy process for you.