UserInertialStarts
OEFastROCSOrientation::UserInertialStarts allows custom
coordinates to be used as starting positions for optimization. Using the
4TMN ligand from the
Introduction as an example,
optimization can be done at the atom highlighted in green below:
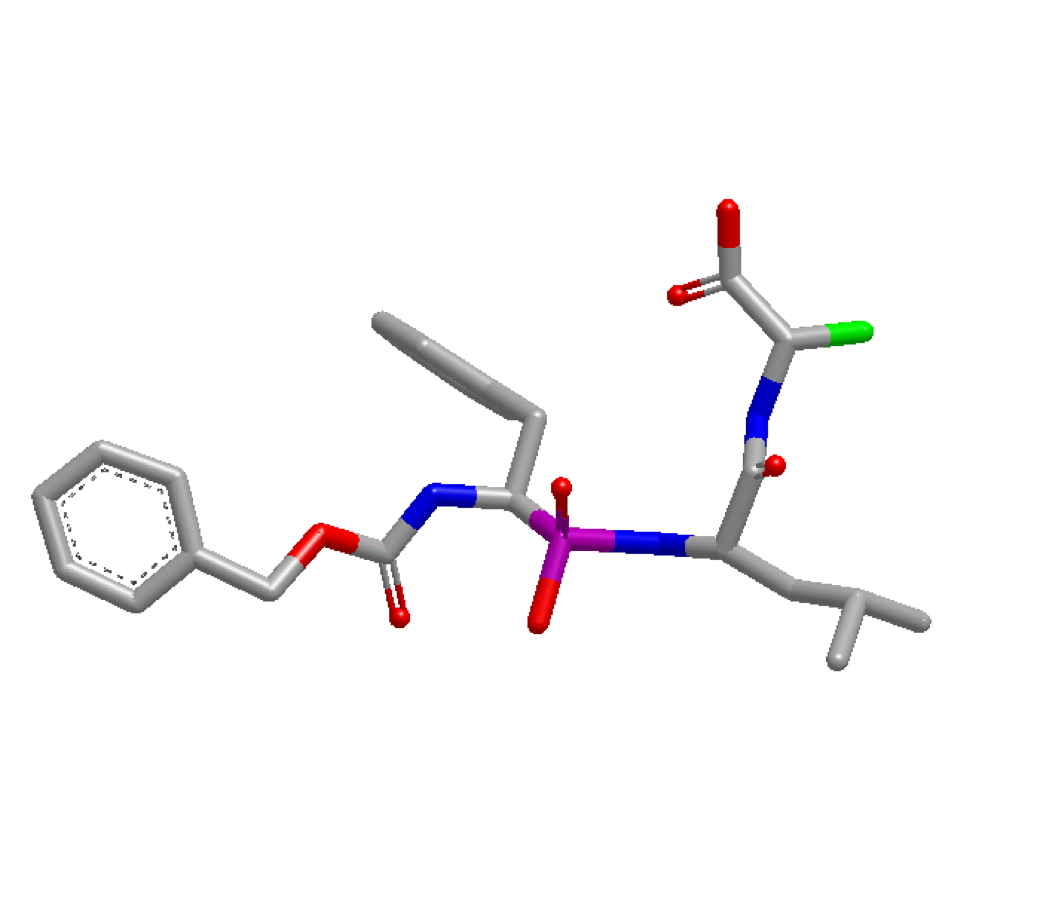
4TMN Ligand with atom 34 highlighted green
Before running the program, the index of the desired atom needs to be identified. This can be accomplished by loading the molecule in VIDA and turning on atom indices. In this example, the atom’s index is identified as idx 34. With this information, the x,y,z coordinates of atom idx 34 are pulled via the code snippet below. These coordinates are then used as user-defined starting coordinates:
float coords[3 * query.GetMaxAtomIdx()];
unsigned int atomIdx = 34;
query.GetCoords(coords);
float c[3];
for(unsigned int k = 0; k < 3; ++k)
c[k] = coords[atomIdx*3 + k];
To use the OEFastROCSOrientation::UserInertialStarts
method, set
OEShapeDatabaseOptions::SetInitialOrientation and then
input the starts
vector using OEShapeDatabaseOptions::SetUserStarts:
opts.SetInitialOrientation(OEFastROCSOrientation::UserInertialStarts);
opts.SetUserStarts(c, 1);
The first argument of
OEShapeDatabaseOptions::SetUserStarts is the
startsCoords array created previously. This must be of the type
float array. The second argument is the number of user-defined
starting coordinates to optimize.
In this example, only 1 user-defined starting point has been set. There is no limit to the number of user-defined starts however, it should be noted that performance is inversely proportional to the number of starts being optimized.
To check that the starts have been set correctly, query
OEShapeDatabaseOptions::GetInitialOrientation and
OEShapeDatabaseOptions::GetNumUserStarts:
if(opts.GetInitialOrientation() == OEFastROCSOrientation::UserInertialStarts)
{
unsigned int numStarts = opts.GetNumUserStarts();
OEThrow.Info("This example will use %u starts", numStarts);
}
The output from the modified C++ script now looks like this:
Opening database file 3tmn_lig.sdf ...
This example will use 1 starts
Searching for 4tmn_lig.sdf
Score for mol 0(conf 0) 0.289689 shape 0.358462 color
Score for mol 0(conf 0) 0.320501 shape 0.098301 color
Score for mol 0(conf 0) 0.283724 shape 0.012400 color
Score for mol 0(conf 0) 0.145704 shape 0.023627 color
Download code
The fully modified c++ script used in this tutorial can be found here
userinertialstarts.cpp