OEToolkits 2017.Feb
Alternative Starting Points for Shape Overlap Optimization in FastROCS TK
This release of FastROCS TK extends its capability to find the best possible shape match between two conformers by introducing the ability to use custom starting points for the shape search. For some systems, using these alternative starting points can lead to more accurate shape scoring as it enables broader sampling of the molecular volume overlap.
By default, the shape search in FastROCS TK starts from 4 poses of the
database molecule aligned around the inertial axes. With the
UserInertialStarts
option, FastROCS TK translates the database molecule to user-defined
starting coordinates. With the
InertialAtHeavyAtoms option,
the database molecule is translated to each heavy atom of the query molecule.
In both cases, the shape overlap is optimized for 4 unique inertial poses
at each location.
The images below illustrate a shape search between caffeine query and a naphthalene derivative.
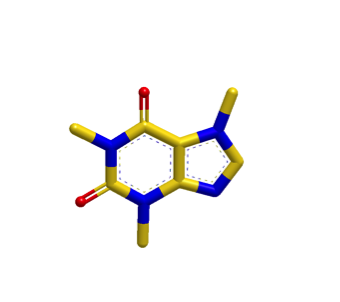
|
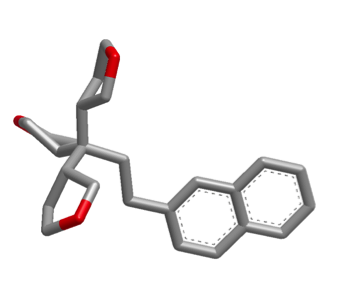
|
The default shape search results in a poor overlap between the two molecules
(left). When the new
InertialAtHeavyAtoms
option is used, the resulting overlap is much more favorable (right).
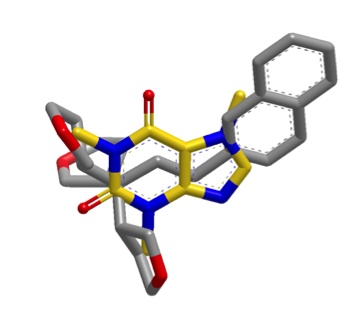
|
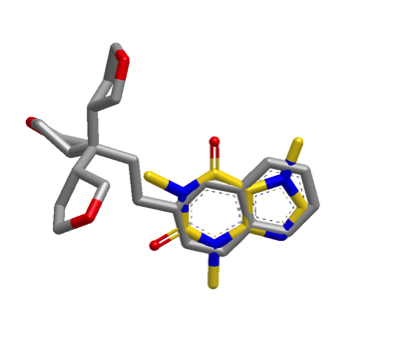
|
In general, these options provide the ability to fine-tune FastROCS TK searching based on specific criteria.
Charging Engine
A new charging function, OEAssignCharges,
has been added. This function introduces the concept of a
charge engine,
a flexible, open-ended
options class that defines a charging method. Charge engines have been
written for every currently supported charging method as well as the new method
OEAM1BCCELF10Charges.
New Maximum Common Substructure (MCS) Similarity Capability
A new class, OEMCSFragDatabase, has been
added that allows pairwise MCS similarity scores to be computed for a
query molecule against a set of indexed target structures. The indexing
algorithm uses a fragmentation approach to reduce the computational
requirements of traditional NxN structure comparisons for MCS
scoring. Tanimoto and Tversky similarity scoring functions are
provided, as well as the ability to implement custom scoring
functions.
Protein Preparation Update
In the 2016.Oct release, new functionality for hydrogen placement was added as part of a protein preparation workflow. This new functionality has been significantly improved.
A new scoring system, MMFF-NIE (“MMFF Neighbor Interaction Energies”),
has been added to OEPlaceHydrogens
that significantly improves optimization of hydrogen bonding
networks and clash avoidance.
In addition, significant improvements have been made in the placement of hydrogens around metal complexes. In the following example, cysteine sulfurs are now properly perceived as deprotonated in the zinc finger domain of the nucleocapsid protein NCp8 (PDB: 2A51).
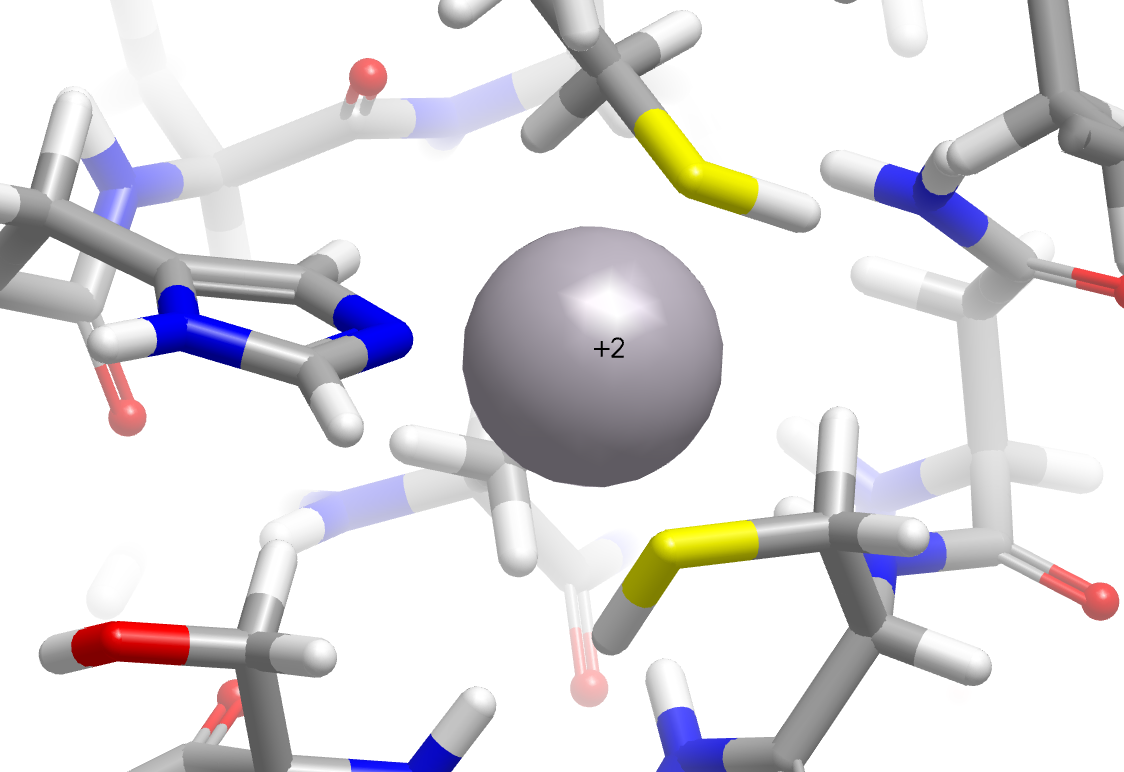
|
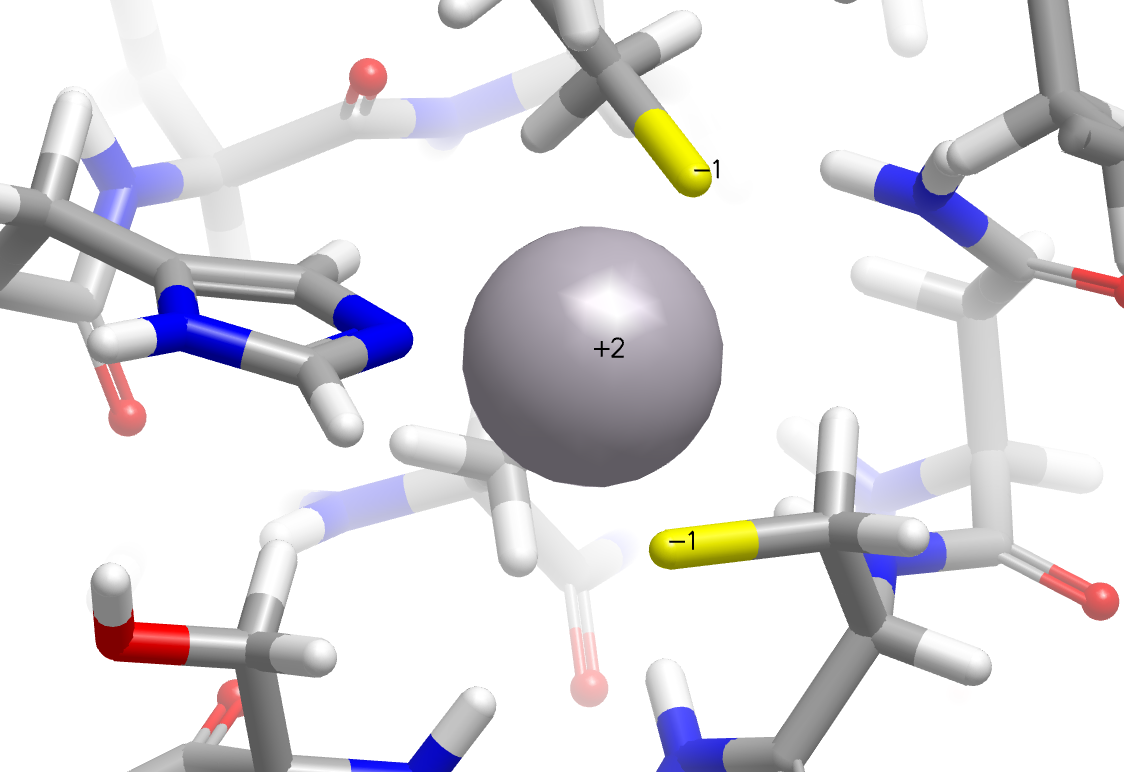
|
General Notices
For Python, the new environment variable
OE_PIP_ARCHcan be set to override the Python package download.OE_ARCHnow only applies to application installation. This should mitigate conflicts between Python and application package installation.On Linux, Python
single-builddistributions are now installed by default when using the meta-installer viapip install openeye-toolkits. If a problem is encountered with thesingle-buildpackages, users can set the environment variableOE_PIP_ARCH=oldbefore thepip installstep.The Java wrappers are now built using Java 1.8 in 1.6 compatibility mode. Users should see no impact.
The 2017.Feb release fully supports macOS Sierra 10.12.
Python 3.4 on Windows is no longer supported.
Python 3.6 will be supported in the 2017.Jun release.
Note
The default Python version installed under Anaconda/Miniconda is
Python 3.6. Until the 2017.Jun release is available, users should
override the default Python version with conda create python=3.5 ...
during the conda environment creation step.
Note
OpenEye continues to monitor and collect feedback from our customers on the need to support Python 2 wrappers. Our current plan is to phase out support for Python 2 on Windows by the 2017.Oct release and phase out support on other platforms at a later date. As this is a substantial change for our customers, we are willing to help with migration. Please contact support@eyesopen.com for more details.