Release Highlights 2022.1
Enhanced Stereo Information Support
The 2022.1 release adds support for enhanced stereochemistry designations in molecules,
defining relationships between stereocenters. Enhanced stereochemistry
information on any molecule is honored by OEFlipper
during stereo enumeration when asked to do so by setting appropriate options. Enhanced
stereochemistry support has also been extended to both the OMEGA and
the Flipper applications.
Support for CXSMILES as a valid molecular input format via
OEFormat::CXSMILES has been added. CXSMILES is specifically
intended to support the import of Enamine download structure files
containing enhanced stereochemistry designations. While many other features are possibly
provided in the CXSMILES appendix information, only the enhanced stereochemistry group
information is supported at this time. Please contact support@eyesopen.com with specific needs
for additional CXSMILES features to allow prioritization in future releases.
OMEGA and Flipper applications can now accept molecule files in CXSMILES format.
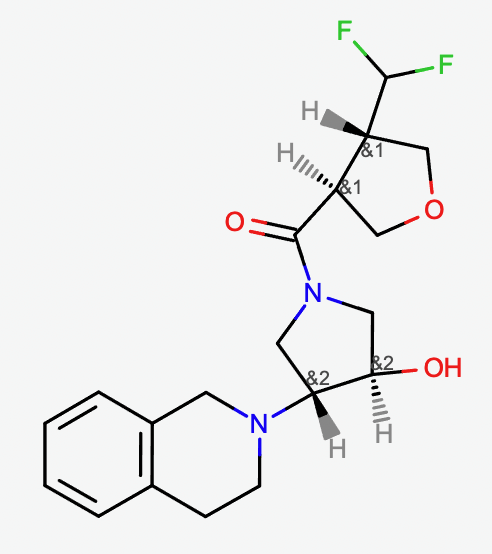
Molecule with enhanced stereo designation. The molecule has 16 stereoisomers when stereocenters are treated independently and only 4 stereoisomers when the enhanced stereo is enabled.
Sheffield Solvation Model for Proteins
The simple two parameter Sheffield Solvation Model, as introduced by Grant et al. ([Grant-2007] ) for calculating the electrostatic component of molecular solvation energy, is a semi-empirical model that performs well for small molecules and requires few computational resources. This model does not work well for macromolecules like proteins and complexes, where many of the atoms are hidden or only partially exposed to the solvent.
A modified three parameter Sheffield Solvation Model is parameterized and implemented in this release,
where solvent exposure is treated by correcting the effective atomic radii with the introduction of
a third additional parameter. The new model is available as part of the
OESheffield API in the OEFF toolkit. The model is also available for use
through the OEFixedProteinLigandOptimizer and the
OEFlexProteinLigandOptimizer APIs in the SZYBKI toolkit, and the OptLigandInDU
and OptimizeDU applications in SZYBKI.
Electrostatic solvation energies for protein conformers are calculated using both the new Sheffield Solvation Model and the Poisson–Boltzmann (PB) model, and the difference in solvation energies between conformers of the same protein is compared between the two models. A set of 232 NMR proteins and peptide structures were used for this study that produced 41491 solvation energy differences between pairs of conformations. The results are compared in the image below.
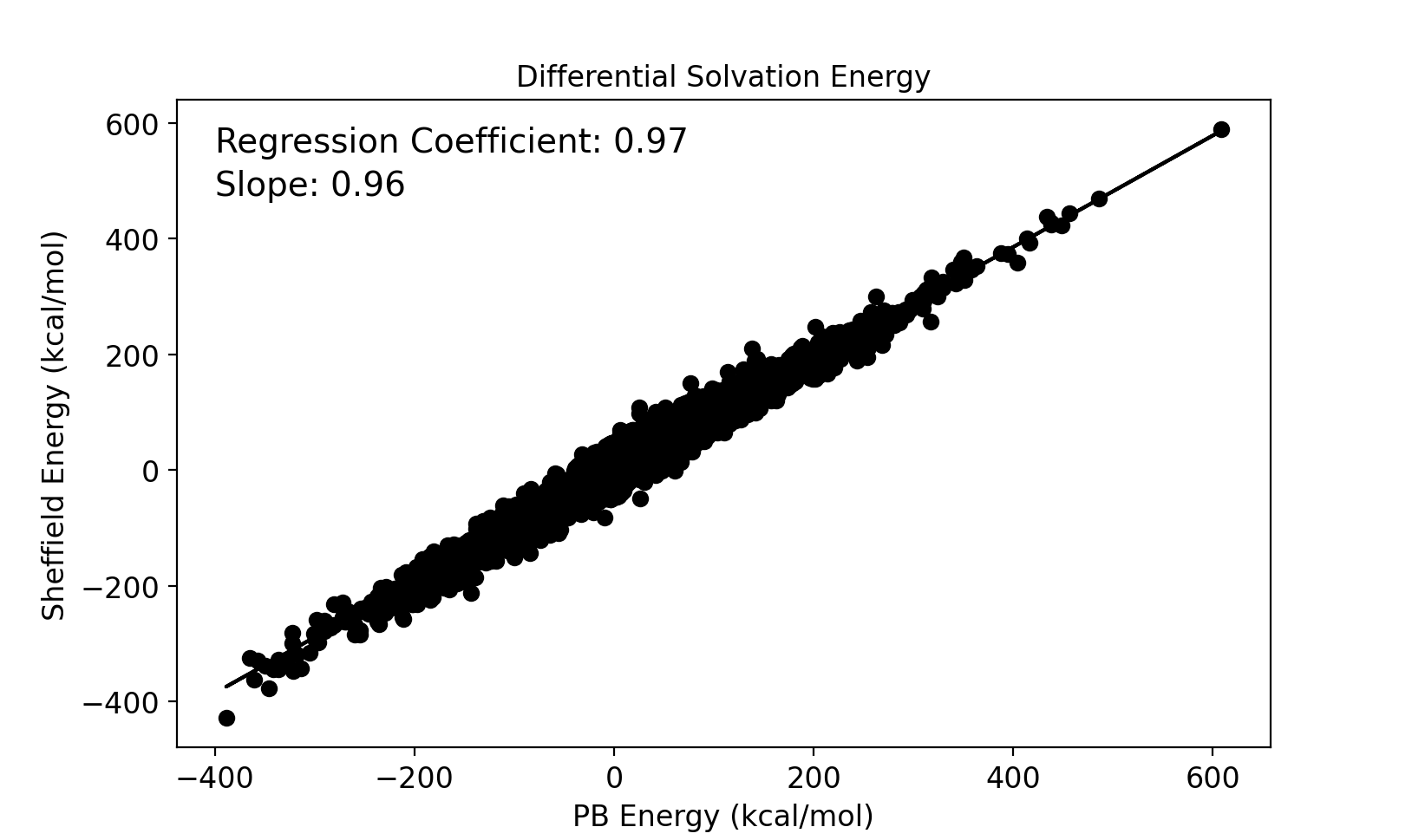
Comparison between Sheffield and PB of differential electrostatic solvation energies between protein conformers.
Spruce Filter and Design Unit Validation
New functionality for preprocessing inputs and validation of the
output Design Units has been added to Spruce TK in the form of two preliminary API classes OESpruceFilter and
OEValidateDesignUnit.
These classes are also used by the SPRUCE application to check and fix, if possible, inputs and to validate the resulting outputs.
OESpruceFilter performs a standardization and a filter check structures to use before prepping
this structure in SPRUCE. It automatically checks and fixes invalid component names, invalid residue
states,and invalid metal bonds. It also checks for invalid CRYST1 from the header file, low overlap between
the electron density map and the structure, invalid sequence alignment, blank chain IDs, and invalid
alternate locations. If the structure does not pass these tests, Spruce Filter will fail the structure.
The OEValidateDesignUnit function performs a check on the design units generated by
SPRUCE. It will report if the provided DU contains partial side chains, missing loops, implicit
hydrogens, incorrect partial charges, termini that are not capped properly, heavy atoms overlapping
with each other, and broken chains.
Multistate Heuristic pKa Model
The multistate heuristic pKa model is an extension of the existing pKa model in OpenEye software. The existing model generates only one probable ionization state based on pKa estimated from about 80 rules. With the development of this multistate pKa model, we are enhancing the implementation with many more unique functional patterns in the form of pKa rules, identified from OpenEye’s internal pKa database, allowing generation of all ionization states possible at physiological pH.
The model described herein uses SMARTS-based heuristics to assess the pKa of acidic or basic functional groups of molecules. For all atoms of a query molecule, pKa is assessed as it belongs to one of three classes: Acidic (pKa < 6.4), Basic (pKa > 8.4), and Neutral (6.4 < pKa < 8.4). According to the assessed pKa class for each atom, it is either ionized and/or un-ionized at pH 7.4. Any group with Acidic or Basic pKa range will result in one state, ionized or un-ionized and a group with Neutral pKa range will result in both states, ionized and un-ionized. Based on how many such functional groups there are in the molecule, and how many states are generated for them, multiple molecules can be generated for all possible combinations of all possible ionization states of acid/base groups.
The multistate heuristic pKa model is currently only available in Quacpac TK as preliminary
API classes OEMultistatepKaModel and OEMultistatepKaModelOptions.
Supported Platforms
Package
Versions
Linux
Windows
macOS
Python
3.7, 3.8, 3.9, 3.10
RHEL7/8, Ubuntu18/20/20-ARM
Win10/11
10.15, 11, 12
C++
RHEL7/8, Ubuntu18/20/20-ARM
Win10/11 (VS2017/19/22)
10.15, 11, 12
Java
1.8, 11
RHEL7/8, Ubuntu18/20/20-ARM
Win10/11
10.15, 11, 12
C#
Win10/11 (VS2017/19/22)
General Notices
Support for macOS 12 has been added. macOS 10.14 is no longer supported.
This is the last release to support macOS 10.15.
A GCC 7.3 C++ toolkit package has been added for RHEL7.
Support for Windows 11 has been added.
Support for Visual Studio 2022 has been added.
Support for Apple M1 architecture has been added for C++ and Python toolkits.
Support for Linux ARM architectures has been added.
This is the last release to support Ubuntu 18.04. Support for Ubuntu 22.04 will be added in the next release.
For detailed release notes, see Version 2022.1.0 Release Notes.