Reactions
Reaction processing in OEChem TK is divided into two categories: unimolecular
reactions and library generation.
Unimolecular reactions are useful for (although not limited to) normalization
reactions.
The OEUniMolecularRxn class in OEChem TK
applies chemical transformations to individual molecules.
Reactions can also be used to generate combinatorial libraries using OEChem TK.
Both ‘clipping’ and reaction based enumeration can be achieved using the
OELibraryGen class.
Figure: OEUniMolecularRxn and
Figure: OELibraryGen demonstrate
the difference between the operation of
OEUniMolecularRxn and the operation of
OELibraryGen.
OEUniMolecularRxn always takes one
starting compound (see Figure: OEUniMolecularRxn)
to which the specified transformation is applied recursively, until no further reactant
pattern are
matched, thereby generating one final product.
See more details about OEUniMolecularRxn class
in section Normalization Reactions.
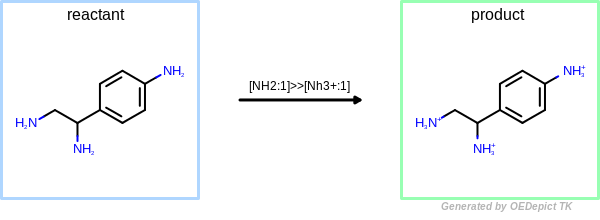
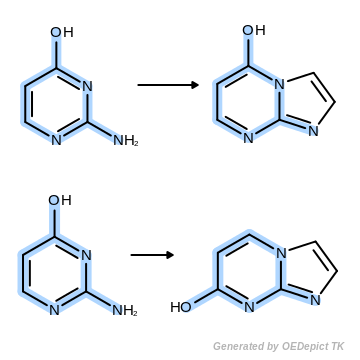
OEUniMolecularRxn
Example of amine nitrogen protonation using ‘OEUniMolecularRxn’. Transformations are applied to the starting material recursively
OELibraryGen performs transformations independently to
each other, enumerating all possible combinations.
As a result, three different products are generated when the same protonation transformation
is applied to the starting material using OELibraryGen
(see Figure: OELibraryGen).
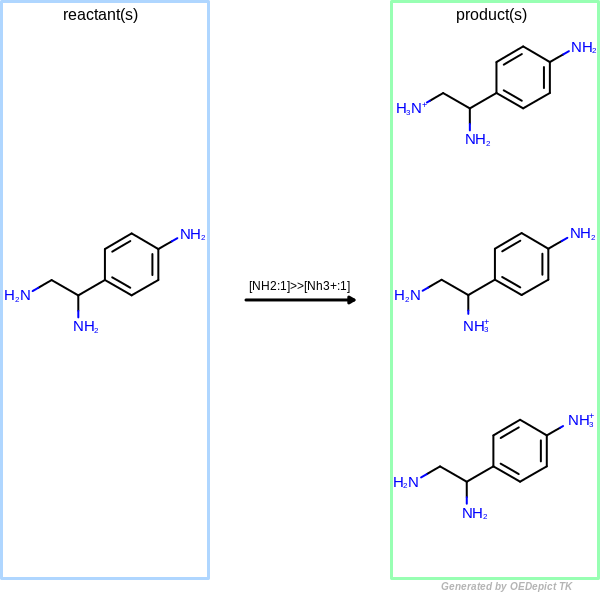
OELibraryGen
Example of amine nitrogen protonation using ‘OELibraryGen’. Transformations are applied to the starting material independently
When OELibraryGen is used to generate combinatorial
libraries (see Figure: Library generation),
all starting material combinations are enumerated to generate products by
considering that any reactant patterns can be mapped to any
starting materials multiple times.
See more details about OELibraryGen class in section
Library Generation.
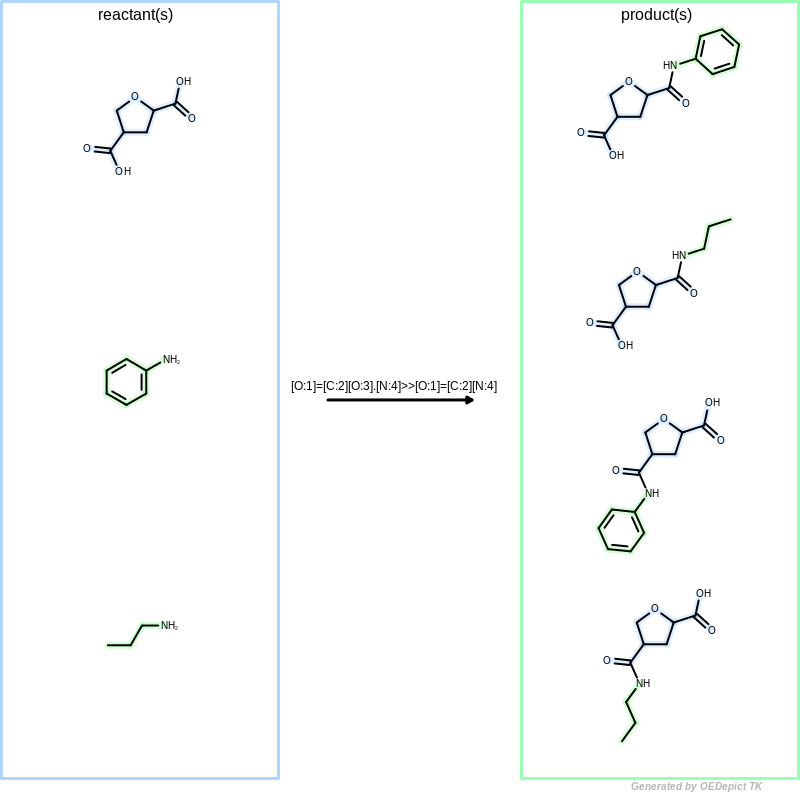
Library generation
Example of library generation. ‘OELibraryGen’ enumerates all possible reacting pattern combinations to generate products.
Reactions are represented in OEChem TK as query molecules ( OEQMolBase ).
Sets of chemical transform operations are derived from reaction molecules by
differences between the reactant and product patterns and in the reaction
molecule.
For example, atoms and bonds that appear in the reactant pattern, but are
absent in the product pattern are destroyed.
Atoms and bonds that appear in the product pattern but not in the reactant
pattern are created.
Atoms are tracked between reactants and products by atom maps.
Atom maps are stored and retrieved using
the SetMapIdx and
the GetMapIdx methods.
Product atoms that have the same map index as reactant atoms originate from
their reactant counterpart.
Reactions are completely defined by a fields in a query molecule.
Reaction molecules can be created from virtually any reaction file format
(SMIRKS, MDL RXN, etc.), and can even be constructed programmatically.
Normalization Reactions
The OEUniMolecularRxn class is designed to
apply a set of transformations defined by a reaction to exactly one reactant
molecule.
All possible transformations are applied to the initial set of atoms and bonds of
an input molecule.
For example, a reaction that affects a particular type of functional group will
be automatically applied twice to a bi-functional molecule.
The number of transformations applied by the
OEUniMolecularRxn class is limited in order
to prevent infinite loops.
Consider a hypothetical reaction that methylates a methyl group.
If a methyl group added in a reaction were allowed to react again, the methyl
groups of a molecule would be methylated ad infinitum.
The first protection against infinite loops provided by
OEUniMolecularRxn is that only original
atoms and bonds of the input molecule are allowed to react.
Atoms and bonds created by a reaction are excluded from involvement in further
reactions.
A more subtle source of potential infinite loops are reactions where product
atoms still match the reactant pattern after they have been involved in a
chemical transformation.
The OEUniMolecularRxn class allows a set of
atoms that match a reactant pattern to react only once.
The following code demonstrates the use of the
OEUniMolecularRxn class.
OEUniMolecularRxn in this case is initialized
using a SMIRKS pattern.
The example reaction (see Figure: Protonation) protonates and charges
an amine nitrogen.
The reaction demonstrates the protection mechanisms in place to prevent
under-specified reactions from causing infinite loops.
The product in Figure: Protonation still matches the reactant pattern,
however, subsequent reactions terminate when no unreacted atoms are identified.
Listing 1: Normalization
#include <openeye.h>
#include <iostream>
#include <oechem.h>
using namespace std;
using namespace OEChem;
int main()
{
OEUniMolecularRxn umr("[NH2:1]>>[Nh3+:1]");
OEGraphMol mol;
OESmilesToMol(mol, "NCCN");
umr(mol);
cout << "smiles = " << OEMolToSmiles(mol) << endl;
return 0;
}
The output of Listing 1 is the following:
smiles = C(C[NH3+])[NH3+]
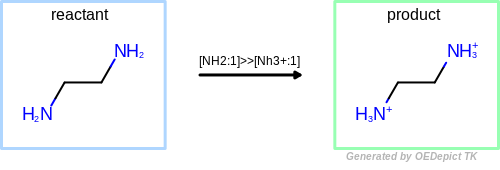
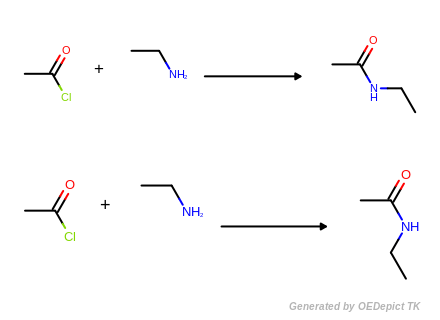
Protonation
Example of amine nitrogen protonation.
Library Generation
The OELibraryGen was designed to give programmers a
high degree of control when applying chemical transformations.
It was also designed for efficiency. Potentially costly preprocessing is performed
once before transformations can be carried out.
The relative setup cost of a OELibraryGen instance may
be high, and the memory use large as preprocessed reactants are stored in memory.
Subsequent generation of products, however, is very efficient because setup
costs are paid in advance.
The OELibraryGen class serves a dual purpose of
managing sets of preprocessed starting materials, and storing a list of chemical
transform operations defined by a reaction molecule.
Chemical transform operations are carried out on starting materials.
Starting materials provide most of the virtual matter that goes into
making virtual product molecules.
The OELibraryGen class provides an interface to
associate starting materials with reactant patterns using the
SetStartingMaterial and
AddStartingMaterial methods.
These methods associate starting materials to reactant patterns using the index
(reactant number) of the pattern.
In a SMIRKS pattern the first reactant (reactant number zero) is the furthest
reactant on the left.
Disconnected reactant patterns may be grouped into a single component using
component level grouping in SMIRKS, denoted by parentheses.
Initialization with SMIRKS
OELibraryGen objects can be directly initialized
with a SMIRKS pattern or an OEQMolBase object.
See example codes in Listing 2 –
Listing 5.
A boolean argument is used to specify whether the SMIRKS string should be
interpreted using strict SMIRKS semantics. See the SMIRKS definition
describing the strict SMIRKS semantics.
Passing a boolean value of false to the second method argument will relax
the strict SMIRKS restrictions.
The Init function returns false,
if OELibraryGen can not be initialized.
OELibraryGen objects can also be initialized from
an MDL reaction query file, for more details and examples see section
MDL Reaction Query File.
Starting Materials
The SetStartingMaterial and
AddStartingMaterial methods
are used to initialize the starting materials corresponding to a reaction component
(reactant).
The declaration of these functions are:
unsigned int SetStartingMaterial(OEIter<OEMolBase>&, unsigned int reacnum, bool umatch=true);
unsigned int AddStartingMaterial(OEIter<OEMolBase>&, unsigned int reacnum, bool umatch=true);
unsigned int SetStartingMaterial(OEIterBase<OEMolBase>*, unsigned int reacnum, bool umatch=true);
unsigned int AddStartingMaterial(OEIterBase<OEMolBase>*, unsigned int reacnum, bool umatch=true);
unsigned int SetStartingMaterial(const OEMolBase&, unsigned int reacnum, bool umatch=true);
unsigned int AddStartingMaterial(const OEMolBase&, unsigned int reacnum, bool umatch=true);
An iterator over molecules or a single molecule may be passed as the first argument to these methods. A copy of each starting material is stored internally along with the atom maps obtained by performing a substructure search with the corresponding reactant pattern.
The difference between using SetStartingMaterial and
AddStartingMaterial methods is that
by calling SetStartingMaterial the
starting materials set in prior calls are discarded, while the
AddStartingMaterial methods append
to the list of starting materials set in prior calls.
The second argument specifies the reactant by number, starting with
zero, to which the starting materials correspond. These numbers
correspond with the left to right lexical ordering of reactants in the
SMIRKS. In the Listing 2 example code,
there is only one reactant pattern specified in the SMIRKS string,
therefore 0 is used to add the corresponding starting materials.
The final argument is used to control the pattern matching of the
reactant pattern to the starting material. If the default true value
is used, then only matches that contain a unique set of atoms relative
to previously identified matches are used. If the value is false,
every possible match including those related by symmetry will be used.
Listing 2 and the corresponding
Figure: Non-unique search shows an
example in which two unique products are generated because the
reactant pattern can be mapped twice when a non-unique substructure
search is performed. For more information about substructure searches
see section Substructure Search.
Both the SetStartingMaterial and
the AddStartingMaterial methods
return the number of matches found in the starting materials.
Listing 2: Library generation with non-unique pattern matching
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
int main()
{
OELibraryGen lg;
lg.Init("[N:3][c:4]1[n:2][c:7][c:6][c:5][n:1]1>>[c:6]1[c:7][n:2][c:4]2[n:3]cc[n:1]2[c:5]1");
lg.SetExplicitHydrogens(false);
lg.SetValenceCorrection(true);
OEGraphMol mol;
OESmilesToMol(mol, "c1cnc(nc1O)N");
const auto reactantNo = 0u;
const bool uniqueMatch = false;
const auto matches = lg.AddStartingMaterial(mol, reactantNo, uniqueMatch);
oeout << "number of matches = " << matches << oeendl;
for (OEIter<const OEMolBase> product = lg.GetProducts(); product; ++product)
oeout << "product smiles = " << OEMolToSmiles(product) << oeendl;
return 0;
}
Non-unique search
Example of generating two unique products when performing non-unique pattern matching.
The output of Listing 2 is shown below:
number of matches = 2
product smiles = c1cnc2nccn2c1O
product smiles = c1cn2ccnc2nc1O
Valence Correction Modes
Once a reaction has been defined, and starting materials have been associated with each of the reactant patterns, chemical transformations can be applied to combinations of starting materials. To achieve a chemically reasonable output attention should be given to the mode of valence (or hydrogen count) correction that matches the reaction.
The OELibraryGen class has three possible modes of
valence correction:
the default explicit hydrogen mode (see example in section Explicit Hydrogen Handling)
the implicit hydrogen mode (see example in section Implicit Hydrogen Handling)
the automatic valence correction mode (see example in section Automatic Valence Correction)
The valence correction mode must be assigned prior to adding starting materials
to an OELibraryGen instance by calling the
SetExplicitHydrogens and the
SetValenceCorrection methods.
Note that the explicit hydrogen setting in effect modifies the semantics of SMIRKS. If the programmer wishes to implement strict SMIRKS according to the Daylight standard, in full, explicit hydrogens should be set on.
Explicit Hydrogen Handling
The default mode for valence correction and SMIRKS interpretation is to emulate the Daylight Reaction Toolkit. Hydrogen counts are adjusted using explicit hydrogens in SMIRKS patterns. Reactions are carried out using explicit hydrogens, and valence correction occurs when explicit hydrogens are added or deleted as defined by a reaction. The following example demonstrates strict SMIRKS and explicit hydrogen handling.
Listing 3: Library generation using explicit hydrogens
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
int main()
{
OELibraryGen libgen("[O:1]=[C:2][Cl:3].[N:4][H:5]>>[O:1]=[C:2][N:4]");
OEGraphMol mol;
OESmilesToMol(mol, "CC(=O)Cl");
unsigned int reactantNo = 0u;
libgen.SetStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "NCC");
reactantNo = 1u;
libgen.SetStartingMaterial(mol, reactantNo);
for (OEIter<const OEMolBase> product = libgen.GetProducts(); product; ++product)
oeout << "product smiles = " << OEMolToSmiles(product) << oeendl;
return 0;
}
The output of Listing 3 is shown below:
product smiles = CCNC(=O)C
product smiles = CCNC(=O)C
The program generates two products, each of them corresponds to the equivalent protons attached to the amine (see example in Figure: Hydrogen handling). If a unique set of products is desired, canonical smiles strings may be stored for verification that products generated are indeed unique.
Hydrogen handling
Example of strict SMIRKS reaction handling with explicit hydrogen
Implicit Hydrogen Handling
The following code demonstrates how the same basic reaction given in the previous example can be carried out using the implicit hydrogen correction mode. Notice that no explicit hydrogens appear in the reaction. Instead, the SMARTS implicit hydrogen count operator appears on the right hand side of the reaction and is used to assign the implicit hydrogen count of the product nitrogen.
Listing 4: Library generation using implicit hydrogens
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
int main()
{
OELibraryGen libgen("[O:1]=[C:2][Cl:3].[N:4]>>[O:1]=[C:2][Nh1:4]");
libgen.SetExplicitHydrogens(false);
OEGraphMol mol;
OESmilesToMol(mol, "CC(=O)Cl");
unsigned int reactantNo = 0u;
libgen.SetStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "NCC");
reactantNo = 1u;
libgen.SetStartingMaterial(mol, reactantNo);
for (OEIter<const OEMolBase> product = libgen.GetProducts(); product; ++product)
oeout << "product smiles = " << OEMolToSmiles(product) << oeendl;
return 0;
}
The output of Listing 4 is the following:
product smiles = CCNC(=O)C
The reaction is written to work with implicit hydrogens (using the lowercase
‘h’ primitive), and the OELibraryGen instance
is set to work in implicit hydrogen mode using the
SetExplicitHydrogens
method.
The SetExplicitHydrogens
method sets the hydrogen handling mode for the OELibraryGen
instance.
OELibraryGen instances are constructed by default
with the explicit hydrogen mode set to true.
If the value is true, the OELibraryGen instance will add
explicit hydrogens to reactant molecules when they are initialized using either the
SetStartingMaterial or the
AddStartingMaterial methods.
If the value is false, then the SetStartingMaterial
and the AddStartingMaterial
methods will suppress any explicit hydrogens in the reactant molecules, and simply
retain the implicit hydrogen counts for remaining non-hydrogen atoms.
Note that the explicit hydrogen setting in effect modifies the semantics of SMIRKS. If the programmer wishes to implement strict SMIRKS according to the Daylight standard, in full, explicit hydrogens should be set on.
Automatic Valence Correction
The final example demonstrates automatic valence correction with implicit hydrogen mode. The automatic valence correction attempts to add or subtract implicit hydrogens in order to retain the valence state observed in the starting materials.
OELibraryGen instances are constructed by default
with the valence correction mode being turned off.
It can be turned on by passing a boolean true value to
SetValenceCorrection
method of OELibraryGen
(see example in Listing 5).
When valence correction mode is enabled, the valence state for each mapped reacting
atom is recorded before any chemical transformations commence.
After the transform operations are complete, OELibraryGen
will attempt to adjust (either increase or decrease) the implicit hydrogen
count on atoms in the product molecule that are involved in the reaction
to match the original valence state of the corresponding reactant atom.
Formal charge is taken into account during the hydrogen count adjustment.
Listing 5: Library generation with automatic valence correction
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
int main()
{
OELibraryGen libgen("[O:1]=[C:2][Cl:3].[N:4]>>[O:1]=[C:2][N:4]");
libgen.SetExplicitHydrogens(false);
libgen.SetValenceCorrection(true);
OEGraphMol mol;
OESmilesToMol(mol, "CC(=O)Cl");
unsigned int reactantNo = 0u;
libgen.SetStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "NCC");
reactantNo = 1u;
libgen.SetStartingMaterial(mol, reactantNo);
for (OEIter<const OEMolBase> product = libgen.GetProducts(); product; ++product)
oeout << "product smiles = " << OEMolToSmiles(product) << oeendl;
return 0;
}
The output of Listing 5 is shown
below:
product smiles = CCNC(=O)C
Note that valence correction in effect modifies the semantics of SMIRKS. Thus, if the programmer wishes to implement strict SMIRKS according to the Daylight standard, in full, valence correction should be turned off.
In general, automatic valence correction is a convenience that allows straightforward reactions to be written in a simplified manner and reduces the burden of valence state bookkeeping. However, reactions that alter the preferred valence state of an atom, oxidation for example, may not be automatically correctable.
Reactant Tracking
The title of a product molecule is generated by concatenating reactant molecule
titles with a delimiter or separator inserted between them.
The default separator is the underscore character ‘_’.
The title separator string may be changed by calling the
SetTitleSeparator method
(see example in Listing 6).
Listing 6: Tracing reactants
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
int main()
{
OELibraryGen libgen("[O:1]=[C:2][Cl:3].[N:4]>>[O:1]=[C:2][N:4]");
libgen.SetExplicitHydrogens(false);
libgen.SetValenceCorrection(true);
unsigned int numProducts = 1u;
unsigned int numMatches = 0u;
OEGraphMol mol;
OESmilesToMol(mol, "CC(=O)Cl acetyl chloride");
unsigned int reactantNo = 0u;
numMatches += libgen.AddStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "c1ccccc1C(=O)Cl benzoyl chloride");
numMatches += libgen.AddStartingMaterial(mol, reactantNo);
numProducts *= numMatches;
mol.Clear();
OESmilesToMol(mol, "NCC ethanamine");
numMatches = 0u;
reactantNo = 1u;
numMatches += libgen.AddStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "OCCN 2-aminoethanol");
numMatches += libgen.AddStartingMaterial(mol, reactantNo);
numProducts *= numMatches;
oeout << "Number of products = " << numProducts << oeendl;
libgen.SetTitleSeparator("#");
for (OEIter<const OEMolBase> product = libgen.GetProducts(); product; ++product)
{
const std::string smi = OEMolToSmiles(product);
oeout << "product smiles = " << smi << " " << product->GetTitle() << oeendl;
}
return 0;
}
If every reactant added to an OELibraryGen instance
has a unique identifier or name, then the title of a generated product unambiguous
determines from which reactants the product was constructed.
The output of Listing 6 is shown below:
Number of products=4
product smiles = CCNC(=O)C acetyl chloride#ethanamine
product smiles = CC(=O)NCCO acetyl chloride#2-aminoethanol
product smiles = CCNC(=O)c1ccccc1 benzoyl chloride#ethanamine
product smiles = c1ccc(cc1)C(=O)NCCO benzoyl chloride#2-aminoethanol
Both SetStartingMaterial and
AddStartingMaterial methods
return the number of substructure matches detected in the compounds(s) added to
OELibraryGen.
Because OELibraryGen enumerates all possible reactant
combinations to generate products, the number of products that will be generated
can be calculated by multiplying the number of reactant matches.
In the example in Listing 6, there are four
expected products, because only one substructure match is detected for each reactant.
Atom Mapping
The atom mapping defines which atoms and bonds remain the same and which are altered on the reactant and products sides of the reaction.
Atoms that appear in both sides of the reaction are left intact.
Atoms and bonds that appear in the product pattern(s) but not in the reactant pattern(s) are created.
Atoms and bonds that disappear in the reactant pattern(s) but not in the product pattern(s) are deleted. When an atom is deleted all of its unmapped explicit hydrogens are automatically removed.
Unmapped reactant atoms are deleted only in strict semantics.
The user should carefully consider whether to add a mapping or to leave unmapped an atom in the reaction. Consider, for example, the first reactant pattern in Figure: Incorrect reaction mapping (top) that can be mapped both to aldehydes or ketones. Because the carbon atom of the carbonyl (mapping index 2) appears in both side of the reaction, it remains intact during the reaction with all of its neighbor atoms that are not described in the reactant pattern. As a result, when the reactant pattern is mapped to an acyl chloride (see Figure: Incorrect reaction mapping (bottom)) the chlorine atom is still bonded to the carbon in the generated product.
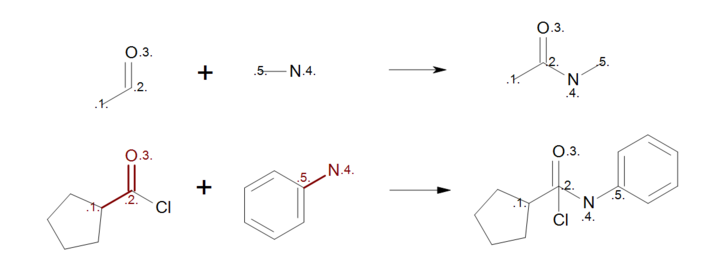
Incorrect reaction mapping
Example of incorrect reaction mapping; (top) reaction definition, (bottom) product generation.
By removing the atom mapping from the carbon atom of the carbonyl (Figure: Correct reaction mapping (top)), the carbon atom will be created on the product side during the product generation. As a result, when the reactant pattern is mapped to an acyl chloride (see Figure: Correct reaction mapping (bottom)), the bond between the carbon and the chlorine atom is broken.
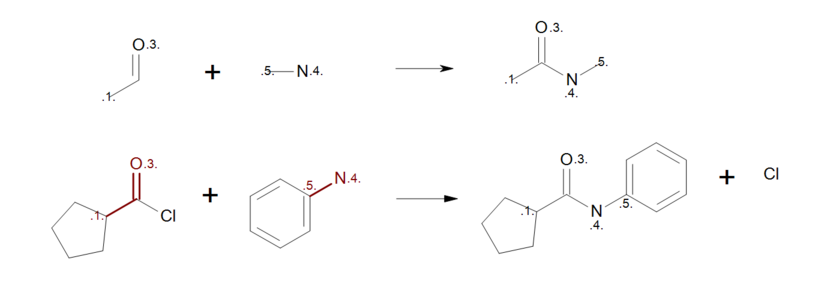
Correct reaction mapping
Example of correct reaction mapping; (top) reaction definition, (bottom) product generation.
The SetRemoveUnmappedFragments
method of OELibraryGen controls whether to keep
or remove the unmapped components on the product side after the reaction is
executed.
When it is enabled, by calling
SetRemoveUnmappedFragments
with the boolean value of ‘true’, any component on the product side that has no
mapped atom is automatically removed.
In case of the example in Figure: Correct reaction mapping (bottom), this means
deleting the isolated chlorine atom.
The default behavior is to keep all components.
Figure: Incomplete reaction (top) shows an example, where not all ring
atoms in the reactants are mapped.
During the product generation, the unmapped atoms on the reactant side are deleted,
while the unmapped atoms on the products side are created.
As a result, ring substitutions are removed from the generated product in all
positions where atom mapping is missing (see generated product and the removed
ring substitution in Figure: Incomplete reaction (bottom)).
The unmapped fragment (CCN) can be automatically removed from the final product
by calling
SetRemoveUnmappedFragments
with the boolean value of ‘true’.
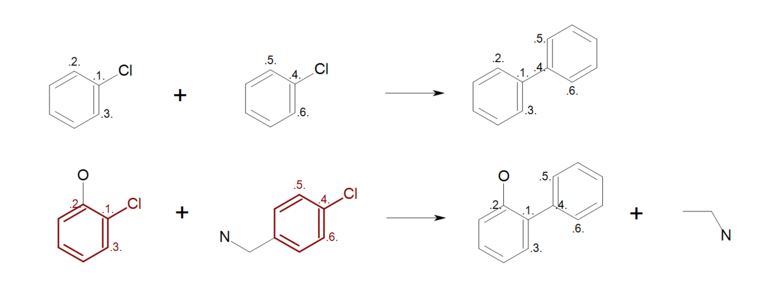
Incomplete reaction
Example of incomplete reaction ring mapping; (top) reaction definition, (bottom) product generation.
By mapping all ring atoms in the reactants (see Figure: Complete reaction (top)), all ring substitutions are left intact in the generated product (see Figure: Complete reaction (bottom)).
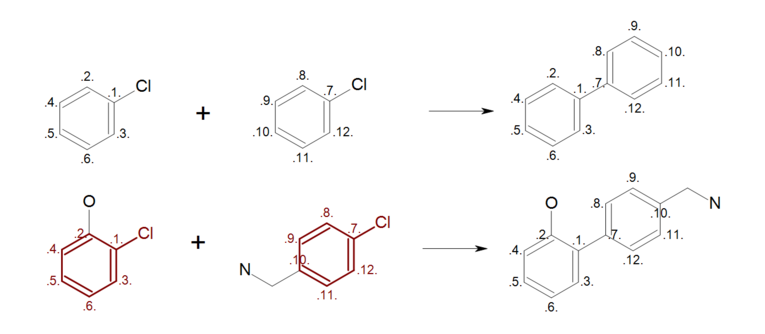
Complete reaction
Example of complete reaction ring mapping; (top) reaction definition, (bottom) product generation.
MDL Reaction Query File
An MDL reaction query file is comprised of a series of structural data with various atom and bond query features to define each reactant and product of a reaction. Mapping indices on the atom blocks define which reactant atom becomes which product atom through the reaction.
See section Supported MDL Query Features for the supported MDL query features in OEChem TK and https://discover.3ds.com/ctfile-documentation-request-form for the MDL reaction file format.
Listing 7 shows an example how to initialize an
OELibraryGen object from MDL reaction query.
The reaction defined in the ‘amide.rxn’ file is shown
Figure: MDL query.
Listing 7: Library generation using a MDL reaction query
#include <openeye.h>
#include <oeplatform.h>
#include <oesystem.h>
#include <oechem.h>
using namespace OEPlatform;
using namespace OESystem;
using namespace OEChem;
static void printatoms(const std::string label, OEIterBase<OEQAtomBase>* begin)
{
oeout << label << oeendl;
for (OEIter<const OEQAtomBase> atom = begin; atom; ++atom)
{
oeout << " atom: " << atom->GetIdx() << ' ' << OEGetAtomicSymbol(atom->GetAtomicNum());
oeout << " in component " << atom->GetData<unsigned int>(OEProperty::Component);
oeout << " with map index " << atom->GetMapIdx() << oeendl;
}
}
int main()
{
oemolistream rfile("amide.rxn");
OEQMol reaction;
// reading reaction
const auto opt = OEMDLQueryOpts::ReactionQuery | OEMDLQueryOpts::SuppressExplicitH;
OEReadMDLReactionQueryFile(rfile, reaction, opt);
printatoms("Reactant atoms:", reaction.GetQAtoms(OEAtomIsInReactant()));
printatoms("Product atoms :", reaction.GetQAtoms(OEAtomIsInProduct()));
// initializing library generator
OELibraryGen libgen;
libgen.Init(reaction);
libgen.SetExplicitHydrogens(false);
libgen.SetValenceCorrection(true);
// adding reactants
OEGraphMol mol;
OESmilesToMol(mol, "c1ccc(cc1)C(=O)O");
unsigned int reactantNo = 0u;
libgen.AddStartingMaterial(mol, reactantNo);
mol.Clear();
OESmilesToMol(mol, "CC(C)CN");
reactantNo = 1u;
libgen.AddStartingMaterial(mol, reactantNo);
// accessing generated products
for (OEIter<const OEMolBase> product = libgen.GetProducts(); product; ++product)
oeout << "product smiles = " << OEMolToSmiles(product) << oeendl;
return 0;
}
The output of Listing 7 is the following:
Reactant atoms:
atom: 0 C in component 1 with map index 1
atom: 1 O in component 1 with map index 0
atom: 2 O in component 1 with map index 2
atom: 3 C in component 2 with map index 4
atom: 4 N in component 2 with map index 3
Product atoms :
atom: 5 C in component 3 with map index 1
atom: 6 N in component 3 with map index 3
atom: 7 C in component 3 with map index 4
atom: 8 O in component 3 with map index 2
product smiles = CC(C)CNC(=O)c1ccccc1
OEReadMDLReactionQueryFile function
reads the MDL reaction query directly into a OEQMolBase
object, which then can be used to initialize an
OELibraryGen object.
OEReadMDLReactionQueryFile(rfile, reaction, opt);
The user can iterate over the reactant and product atoms of a reaction using
the OEAtomIsInReactant and the
OEAtomIsInProduct functors, respectively.
printatoms("Reactant atoms:", reaction.GetQAtoms(OEAtomIsInReactant()));
printatoms("Product atoms :", reaction.GetQAtoms(OEAtomIsInProduct()));
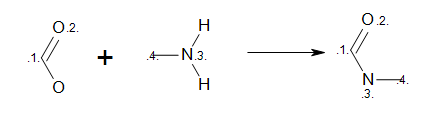
MDL Query
Example of MDL query reaction
The reaction ‘role’ property of an atom can also be accessed with the
GetRxnRole method
of the OEAtomBase class.
The GetMapIdx method returns the
atom map index read from the reaction file.
oeout << " with map index " << atom->GetMapIdx() << oeendl;
An unmapped atom has a map index zero.
For more information about these functions and the OERxnRole
namespace, please refer to the OEChem API manual.
Since the explicit hydrogens are suppressed when the reaction is converted into a query molecule,
const auto opt = OEMDLQueryOpts::ReactionQuery | OEMDLQueryOpts::SuppressExplicitH;
the explicit hydrogens in the starting materials can also be suppressed, yielding faster performance while still generating the same products.
libgen.SetExplicitHydrogens(false);
More details on suppressing hydrogens in a query can be found in section Explicit Hydrogens. It is also recommended to set the valence correction mode on.
libgen.SetValenceCorrection(true);
The MDL reaction query can also be read into a OEMolBase
object (see code snippet below).
In this case, the OEReadMDLReactionQueryFile
function attaches the query features present in the input MDL file to the related
atoms and bonds of the OEMolBase object.
The OEQMolBase object can be subsequently created by
calling the OEBuildMDLQueryExpressions
function with OEMDLQueryOpts::ReactionQuery option.
OEGraphMol reaction;
OEReadMDLReactionQueryFile(rfile,reaction);
// reaction can be manipulated here
OEQMol qreaction;
// build OEQMol with OEMDLQueryOpts::ReactionQuery option
OEBuildMDLQueryExpressions(qreaction,reaction,OEMDLQueryOpts::ReactionQuery);
OELibraryGen libgen;
libgen.Init(qreaction);
The declarations of these functions are:
bool OEReadMDLReactionQueryFile(oemolistream &ifs, OEMolBase& mol);
bool OEReadMDLReactionQueryFile(oemolistream &ifs, OEQMolBase& qmol,
unsigned int opts = OEMDLQueryOpts::ReactionQuery);
bool OEBuildMDLQueryExpressions(OEQMolBase &qmol, const OEMolBase &mol,
unsigned int opts = OEMDLQueryOpts::Default);
The ‘opts’ parameter defines how the MDL query is interpreted when
OEQMolBase object is constructed.
For more details on the various options in the OEMDLQueryOpts
namespace and the MDL query based substructure search, please see chapter
Substructure Search with MDL Queries.
Hydrogens on the Reactant Side
When defining a reaction it is very important to consider not only correct atom mapping but also to anticipate the different kind of starting materials the reactant pattern(s) will match.
At first glance, the reaction shown in Figure: Missing hydrogen (reaction) seems correct, however some of the generated products have carbon atom with incorrect 5 valences see example ‘A’ in Figure: Missing hydrogen (products).
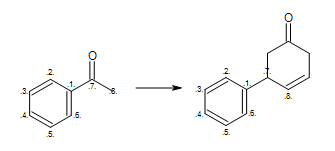
Missing hydrogen (reaction)
Example of missing explicit/implicit hydrogen in reactant (equivalent SMIRKS: [c:2]1[c:3][c:4][c:5][c:6][c:1]1([C:7](=O)[C:8])>>[c:2]1[c:3][c:4][c:5][c:6][c:1]1([C:7]2[C:8]=CCC(=O)C2)
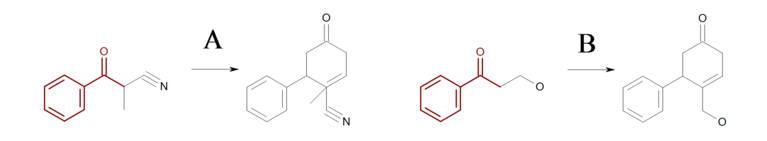
Missing hydrogen (products)
Example of products generated with reaction defined in ‘Missing hydrogen (reaction)’
By adding two explicit (or implicit) hydrogens to the reactant atom with atom map 8
in Figure: Missing hydrogen (reaction),
ensures that only reaction ‘B’
is executed, since the starting material of reaction ‘A’ no longer matches the
reaction pattern.
Note
In order to generate products with correct valences, a mapped reactant atom should have as many as explicit or implicit hydrogens as many as new bonds the atom forms on the product side
Product Kekulization
Prior to returning a generated product, OELibraryGen calls
the OEKekulize and
OEAssignAromaticFlags functions to
determine the Kekulé form and aromaticity of the generated product.
Since OEKekulize requires the correct specification
of the formal charges and implicit hydrogen counts of each atom, the Kekulé form of
a generated product depends on the reaction specification.
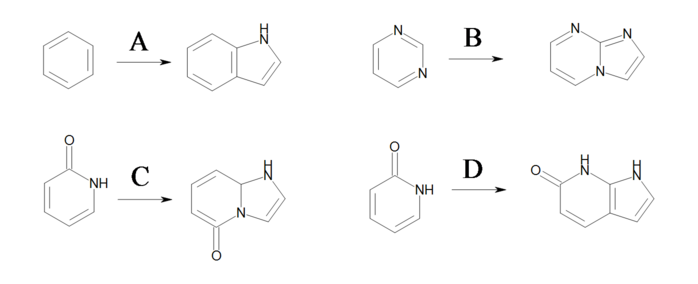
The three reactions shown in Identical transformations describe the same set of transformations, but they generate products with different but valid Kekulé forms. Reactions ‘A’ and ‘B’ in Identical transformations will produce products ‘A’ and ‘B’ in Products, respectively. However, reaction ‘C’ arbitrarily generates either product ‘A’ or its tautomer product ‘B’, since this reaction describes ambiguous Kekulé form for the 5-membered ring in the product.
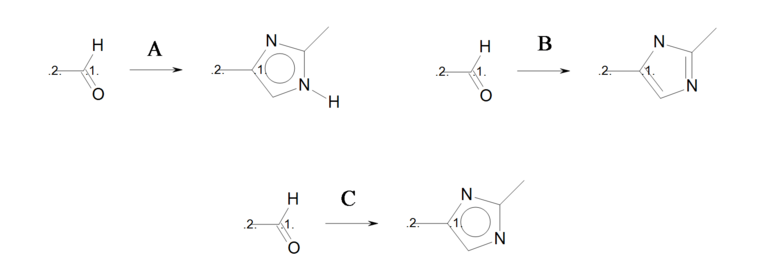
Identical transformations
Three reactions describing the same transformation
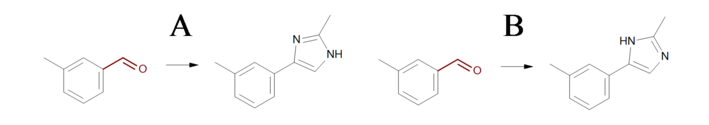
Products
Example of products generated with reactions defined in ‘Identical transformations’
Note
In order to generate products with the desired Kekulé form, either the product has to have only specific bonds or explicit/implicit hydrogens have to be specified to eliminate ambiguities.
Even though the reaction in Reaction defines an
unambiguous Kekulé form, this form is not suited for every product generated by
the reaction (see examples in Products).
In order to ensure that the OELibraryGen generates
products with valid Kekulé form the following steps are performed.
First, Kekulization is performed based on the reaction specification.
If it is unsuccessful, i.e. OEKekulize function
returns false, alternatives are tried by adding and removing implicit hydrogens
from aromatic nitrogens, until a valid Kekulé form is found.
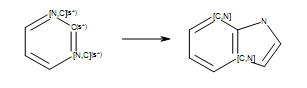
Reaction
Example of reaction with generic atoms in an aromatic ring system (``s*`` = number of non-hydrogen substituents as drawn).
Products
Example of products generated with the reaction shown in previous figure