Stereochemistry Perception
OEChem TK has the ability to store and retrieve stereochemical information for atoms and bonds independent of two or three dimensional coordinates. The current version of OEChem TK supports stereochemistry definitions of handedness around tetrahedral centers, and cis/trans configuration around bonds.
The table below lists the available OEChem TK functions related to stereochemical information.
Name
Description
Set the stereochemistry at the chiral atoms of a molecule, as specified by the molecule’s 3D coordinates.
Set the stereochemistry at the chiral bonds of a molecule, as specified by the molecule’s 3D coordinates.
Assigns the stereochemistry of a molecule from its 3D coordinates.
Clears the bond stereo fields from all bonds of a molecule.
Corrects the wedge/hash bond assignment of the 2D molecule.
Checks whether the wedge/hash bond assignment of the 2D molecule is inconsistent or ambiguous.
Assigns wedge and hash bonds to a connection table from the OEChem TK stereochemistry of each atom of the 2D molecule.
Sets the OEChem TK stereochemistry of each atom of the 2D molecule from the wedge and hash bonds in the connection table.
Sets the OEChem TK stereochemistry for each atom from the MDL stereo parity information.
Perceives the Cahn-Ingold-Prelog descriptor for the given atom.
Sets the internal OEChem TK stereochemistry from the given CIP atom stereo descriptor.
Atom Chirality
The following two code snippets demonstrate how to loop over chiral
atoms using the OEIsChiralAtom functor and
the IsChiral method of
the OEAtomBase class.
for (OEIter<const OEAtomBase> atom = mol.GetAtoms(OEIsChiralAtom()); atom; ++atom)
cout << atom->GetIdx() << ' ' << OEGetAtomicSymbol(atom->GetAtomicNum()) << endl;
for (OEIter<const OEAtomBase> atom = mol.GetAtoms(); atom; ++atom)
{
if (atom->IsChiral())
cout << atom->GetIdx() << ' ' << OEGetAtomicSymbol(atom->GetAtomicNum()) << endl;
}
Warning
When a molecule is imported from a file, its atom and bond chirality are not
automatically perceived, the user has to call OEPerceiveChiral
explicitly.
Atom Stereochemistry
Stereochemistry around atoms can be set and retrieved by the
OEAtomBase::SetStereo and OEAtomBase::GetStereo
methods, respectively.
The OEAtomBase::HasStereoSpecified method returns a boolean value
that indicates whether stereochemical information of a particular type has been
stored for an atom.
OEChem TK currently only supports OEAtomStereo::Tetrahedral
atom stereochemistry class.
Note that the setting and retrieval of atom stereo requires
a vector containing pointers to the neighboring atoms as the first argument.
The handedness value returned from OEAtomBase::GetStereo will
depend on the order of the neighboring atoms as they appear in the
passed vector.
The first neighbor atom in the vector passed to OEAtomBase::GetStereo
is taken as atom number one for the determination of handedness.
Likewise, subsequent atoms in the neighbor atom vector are assigned
sequentially to positions in the handedness definition.
Although, three neighboring atoms are sufficient to determine the
handedness around a trigonal pyramidal or tetrahedral center, either three or four
atoms can be provided to the OEAtomBase::GetStereo function
when requesting a value for tetrahedral chirality.
The following table shows the return values of OEAtomBase::GetStereo
method by passing the neighbor atoms in different order.
neighbor order |
[C@@H](Cl)(Br)N |
[C@H](Cl)(Br)N |
|---|---|---|
H Cl Br N |
right handed |
left handed |
H Br Cl N |
left handed |
right handed |
H Cl N Br |
left handed |
right handed |
… |
… |
… |
Cl Br N |
left handed |
right handed |
Br Cl N |
right handed |
left handed |
Cl N Br |
right handed |
left handed |
Note
The definition of handedness for tetrahedral stereochemistry does not imply chirality around a tetrahedral center, but rather indicates relative positions of neighboring atoms.
The following code sample demonstrates looping over atoms, testing for
atoms that have tetrahedral stereochemistry, and printing out the
value of the tetrahedral stereochemistry (either
OEAtomStereo::RightHanded,
OEAtomStereo::LeftHanded, or
OEAtomStereo::Undefined). See also the input
structure depicted in Figure: Atom stereo.
Listing 1: Accessing atom stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEGraphMol mol;
OESmilesToMol(mol, "[H][C@]1(NC[C@@H](CC1CO[C@H]2CC[C@@H](CC2)O)N)[H]");
for (OEIter<const OEAtomBase> atom = mol.GetAtoms(); atom; ++atom)
{
const bool chiral = atom->IsChiral();
unsigned int stereo = OEAtomStereo::Undefined;
if (atom->HasStereoSpecified(OEAtomStereo::Tetrahedral))
{
vector<OEAtomBase*> vecNbrs;
for (OEIter<OEAtomBase> nbr = atom->GetAtoms() ;nbr; ++nbr)
vecNbrs.push_back(nbr);
stereo = atom->GetStereo(vecNbrs, OEAtomStereo::Tetrahedral);
}
if (chiral || stereo != OEAtomStereo::Undefined)
{
cout << "Atom: " << atom->GetIdx() << " chiral= " << chiral << " stereo= ";
if (stereo == OEAtomStereo::RightHanded)
cout << "right handed";
else if (stereo == OEAtomStereo::LeftHanded)
cout << "left handed";
else
cout << "undefined";
cout << endl;
}
}
return 0;
}
The output of the preceding program is the following:
Atom: 1 chiral=0 stereo=left handed
Atom: 4 chiral=1 stereo=right handed
Atom: 7 chiral=1 stereo=undefined
Atom: 10 chiral=0 stereo=left handed
Atom: 14 chiral=0 stereo=right handed
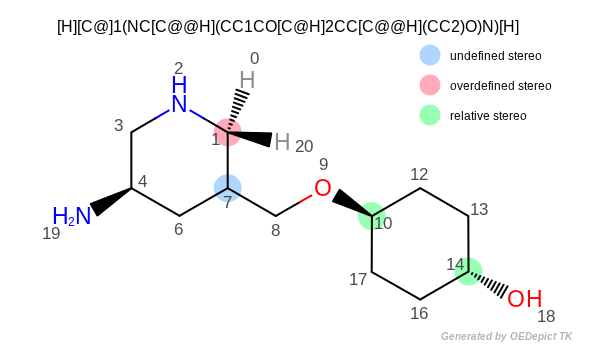
Atom stereo
As mentioned before, tetrahedral stereochemistry (i.e. specified atom stereo) does not imply chirality around a tetrahedral center. For example atoms 1, 10 and 14 in Figure: Atom stereo have specified atom stereo, even though they are non-chiral. Vice versa, atom can be chiral but with unspecified atom stereo configuration (such as atom 7 in Figure: Atom stereo ). Atoms 1 and 6 are non-chiral but they have atom stereo information and they are relative to each other.
The following example shows how to manually set the atom stereo of atom 7 (chiral, undefined)
to left by the OEAtomBase::SetStereo method.
Listing 2: Setting atom stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEMol mol;
OESmilesToMol(mol, "[H][C@]1(NC[C@@H](CC1CO[C@H]2CC[C@@H](CC2)O)N)[H]");
for (OEIter<OEAtomBase> atom = mol.GetAtoms(); atom; ++atom)
{
if (atom->IsChiral() && ! atom->HasStereoSpecified(OEAtomStereo::Tetrahedral))
{
vector<OEAtomBase*> vecAtoms;
for (OEIter<OEAtomBase> neigh = atom->GetAtoms(); neigh; ++neigh)
vecAtoms.push_back(neigh);
atom->SetStereo(vecAtoms, OEAtomStereo::Tetra, OEAtomStereo::Left);
}
}
cout << OEMolToSmiles(mol) << endl;
return 0;
}
The output of the preceding program is the following:
[H][C@@]1([C@@H](C[C@H](CN1)N)CO[C@H]2CC[C@@H](CC2)O)[H]
Just as in OEAtomBase::GetStereo, the vector of neighbor atoms provide
the references about which the handedness is defined.
The first of the integer arguments is the stereochemistry type
(i.e. OEAtomStereo::Tetrahedral (or just OEAtomStereo::Tetra) ,
and the second is the associated value
(i.e. OEAtomStereo::Right or OEAtomStereo::Left).
Bond Chirality
The following two code snippets demonstrate how to loop over chiral
bonds using the OEIsChiralBond functor and
the IsChiral method of
the OEBondBase class.
for (OEIter<const OEBondBase> bond = mol.GetBonds(OEIsChiralBond()); bond; ++bond)
cout << bond->GetBgnIdx() << '=' << bond->GetEndIdx() << endl;
for (OEIter<const OEBondBase> bond = mol.GetBonds(); bond; ++bond)
{
if (bond->IsChiral())
cout << bond->GetBgnIdx() << '=' << bond->GetEndIdx() << endl;
}
Warning
When a molecule is imported from a file, its atom and bond chirality are not
automatically perceived, the user has to call OEPerceiveChiral
explicitly.
Bond Stereochemistry
Stereochemistry around bonds can be specified in a similar fashion to
stereochemistry about atom centers.
The request for stereochemistry of a particular class can be made of an
OEBondBase using the method OEBondBase::HasStereoSpecified.
OEChem TK currently only supports OEBondStereo::CisTrans
bond stereochemistry class that defines whether the configuration
of two atoms around a non-rotatable bond is cis or trans.
If a bond has associated stereochemical data, it can be retrieved
using OEBondBase::GetStereo method.
The following code sample demonstrates a loop over bonds that tests for bonds with associated stereochemistry, and retrieval of whether the neighboring atoms are cis or trans relative to one another. (See also the input structure depicted in Figure: Bond stereo.
Listing 3: Accessing bond stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEGraphMol mol;
OESmilesToMol(mol, "C\\C=C\\C=C/C=CC");
for (OEIter<const OEBondBase> bond = mol.GetBonds(); bond; ++bond)
{
if (bond->HasStereoSpecified(OEBondStereo::CisTrans))
{
for (OEIter<OEAtomBase> atom1 = bond->GetBgn()->GetAtoms(); atom1; ++atom1)
{
if (atom1 != bond->GetEnd())
{
for (OEIter<OEAtomBase> atom2 = bond->GetEnd()->GetAtoms(); atom2; ++atom2)
{
if (atom2 != bond->GetBgn())
{
vector<OEAtomBase*> vecAtoms;
vecAtoms.push_back(atom1);
vecAtoms.push_back(atom2);
const auto stereo =
bond->GetStereo(vecAtoms, OEBondStereo::CisTrans);
cout << "Atoms: " << atom1->GetIdx() << " and ";
cout << atom2->GetIdx() << " are ";
if (stereo == OEBondStereo::Cis)
cout << "cis" << endl;
else if (stereo == OEBondStereo::Trans)
cout << "trans" << endl;
}
}
}
}
}
}
return 0;
}
The output of the preceding program is the following:
Atoms: 0 and 3 are trans
Atoms: 2 and 5 are cis
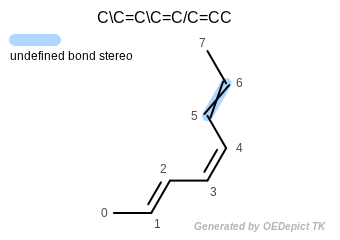
Bond stereo
Note that even though the double bond defined by atoms 4–5–6–7 is depicted as trans (see Figure: Bond stereo) it is still considered undefined, since no stereo information is encoded for this bond in the input SMILES.
The following example shows how to manually set this missing bond stereo to trans
by the OEBondBase::SetStereo method.
Listing 4: Setting bond stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEMol mol;
OESmilesToMol(mol, "C\\C=C\\C=C/C=CC");
for (OEIter<OEBondBase> bond = mol.GetBonds(); bond; ++bond)
{
if (bond->GetOrder() == 2u && ! bond->HasStereoSpecified(OEBondStereo::CisTrans))
{
vector<OEAtomBase*> vecAtoms;
for (OEIter<OEAtomBase> neigh = bond->GetBgn()->GetAtoms(); neigh; ++neigh)
{
if (neigh != bond->GetEnd())
{
vecAtoms.push_back(neigh);
break;
}
}
for (OEIter<OEAtomBase> neigh = bond->GetEnd()->GetAtoms(); neigh; ++neigh)
{
if (neigh != bond->GetBgn())
{
vecAtoms.push_back(neigh);
break;
}
}
bond->SetStereo(vecAtoms, OEBondStereo::CisTrans, OEBondStereo::Trans);
}
}
cout << OEMolToSmiles(mol) << endl;
return 0;
}
The first integer argument of the method OEBondBase::SetStereo
is used to specify the type of stereochemistry (i.e.
OEBondStereo::CisTrans).
The second integer argument specifies the bond stereo configuration (i.e.
OEBondStereo::Cis or OEBondStereo::Trans)
The output of the preceding program is the following:
C/C=C/C=C\C=C\C
CIP Stereo Perception
The following two examples demonstrate how to perceive CIP (Cahn–Ingold–Prelog)
atom and bond stereo information from the molecules created by the SMILES
representation. If the input molecule has 3D coordinates, the OE3DToInternalStereo
function should be called.
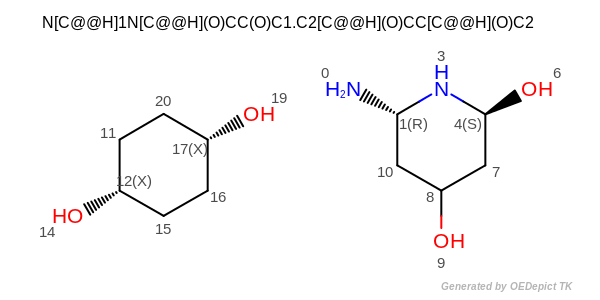
CIP atom stereo
Listing 5: Perceiving CIP atom stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEGraphMol mol;
OESmilesToMol(mol, "N[C@@H]1N[C@@H](O)CC(O)C1.C2[C@@H](O)CC[C@@H](O)C2");
for (OEIter<const OEAtomBase> atom = mol.GetAtoms(); atom; ++atom)
{
const auto cip = OEPerceiveCIPStereo(mol, atom);
if (atom->HasStereoSpecified())
{
cout << "atom " << atom->GetIdx() << " is ";
if (cip == OECIPAtomStereo::S)
cout << "S" << endl;
if (cip == OECIPAtomStereo::R)
cout << "R" << endl;
if (cip == OECIPAtomStereo::NotStereo)
cout << "not a CIP stereo center" << endl;
}
if (cip == OECIPAtomStereo::UnspecStereo)
{
cout << "atom " << atom->GetIdx() << " is ";
cout << "a CIP stereo center without specified stereochemistry" << endl;
}
}
return 0;
}
The OEPerceiveCIPStereo function detects the Cahn-Ingold-Prelog
descriptor of the given atom and returns a value from the
OECIPAtomStereo namespace.
The output of Listing 5 is the following:
(See also the input structure depicted in Figure: CIP atom stereo.
atom 1 is R
atom 4 is S
atom 8 is a CIP stereo center without specified stereochemistry
atom 12 is not a CIP stereo center
atom 17 is not a CIP stereo center
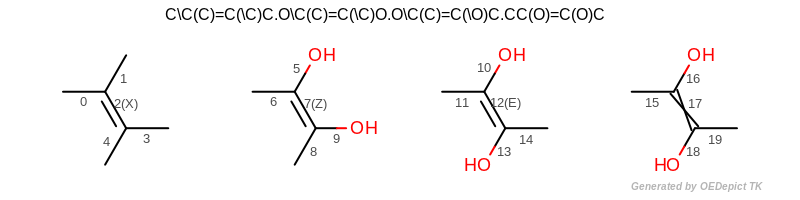
CIP bond stereo
Listing 6: Perceiving CIP bond stereo information
#include <openeye.h>
#include <iostream>
#include <vector>
#include <oechem.h>
using namespace std;
using namespace OESystem;
using namespace OEChem;
int main()
{
OEGraphMol mol;
OESmilesToMol(mol, "C\\C(C)=C(\\C)C.O\\C(C)=C(\\C)O.O\\C(C)=C(\\O)C.CC(O)=C(O)C");
for (OEIter<const OEBondBase> bond = mol.GetBonds(); bond; ++bond)
{
if (bond->GetOrder() == 2u)
{
const auto cip = OEPerceiveCIPStereo(mol, bond);
cout << "bond " << bond->GetIdx() << " is ";
if (bond->HasStereoSpecified())
{
if (cip == OECIPBondStereo::E)
cout << "E" << endl;
if (cip == OECIPBondStereo::Z)
cout << "Z" << endl;
if (cip == OECIPBondStereo::NotStereo)
cout << "not a CIP stereo center" << endl;
}
if (cip == OECIPBondStereo::UnspecStereo)
cout << "a CIP stereo center without specified stereochemistry" << endl;
}
}
return 0;
}
The OEPerceiveCIPStereo function detects the Cahn-Ingold-Prelog
descriptor of the given bond and returns a value from the
OECIPBondStereo namespace.
The output of Listing 6 is the following:
(See also the input structure depicted in Figure: CIP bond stereo.
bond 2 is not a CIP stereo center
bond 7 is Z
bond 12 is E
bond 17 is a CIP stereo center without specified stereochemistry