Pose Prediction¶
Docking is the process of determining the structure of a ligand bound in the
active site of a target protein. OpenEye’s Posit method of pose prediction
consists of choosing the best method to use when docking a particular
ligand to a receptor and then returning the probability that the
docked pose is within 2.0A of the actual pose. In the Docking
Toolkit this is done with the OEPosit class that
takes either a single or multi conformer representation of a ligand
and returns the pose with the highest probability of being correctly
placed in the active site.
Note
Note that this probability is not the probability of binding, rather, the probability that the pose is correct given the ligand actually binds to the receptor.
The methods Posit uses to dock, in order of reliability, are:
MCS Overlay [Hare-2004]
ShapeFit - Shape-guided ligand minimization into the receptor site
Hybrid - hybrid method that uses ligand and protein information
Fred - Standard docking method that uses no ligand information
Posit automatically chooses the best method based on the 2D (graph) and 3D (structure) similarity of the docked ligand to the bound ligand. As a contrived example, if there is no bound ligand in the receptor, the Fred method is used by default.
By default, in addition to the input conformations, Posit generates new conformations during its search and, in some cases, minimizes these structures with the internal force-field.
Note
The selection of methods can be overridden by constructing
an OEPosit with
an OEPositOptions object. See
the OEPositMethod namespace for a list
of available methods.
A note on stereo - isomerisms and chirality¶
Stereo, and notably nitrogen aniline stereo centers, are currently somewhat problematic for POSIT. Many crystal structures have flat geometries for some stereo centers due to time-averaging during data collection. This makes stereo centers appear to have flat geometries in the 3D coordinates.
Because the POSIT algorithm internally expands conformations during the flexible fitting procedure, the full molecule must be labeled with stereo - either in the 3D coordinate sense or the 2D coordinate sense. This means that some pdb ligand structures will unfortunately be failed by the POSIT algorithm.
To get around this, posit can be told to ignore nitrogen stereo when
constructing an OEPosit with an
OEPositOptions object that has the
OEPositOptions.SetIgnoreNitrogenStereo method
set to true.
The following are the recommended ways to supply or prepare molecules for POSIT.
Use OMEGA or the OMEGA toolkit to prepare molecules or expand the stereo chemistry using OEFlipper. POSIT is guaranteed to work correctly if the input molecules have been generated by OMEGA.
Set the OEPositOptions fullConformationalSearch option to be false. See the section OEPositOptions for more details. This method of running POSIT is not recommended as a large portion of the flexible methodology will be deactivated.
Note
Output structures of Posit are not guaranteed to have the same conformations of the input molecule. This is occasionally problematic if the force-field minimizations alter the desired input conformations. This issue will be addressed in a future release of the Docking Toolkit.
POSIT Algorithm¶
POSIT supplies a robust probability that the given pose is reasonable. It is generally recognized that docking and scoring methods have inaccuracies and do not provide a measure that can be compared between different complexes. For example, a docking score from one complex cannot be directly compared to a docking score form another.
POSIT overcomes this by using 2D, 3D and protein-ligand information to generate a probability that the pose has been correctly placed. This probability is independent of how the ligand was actually placed, and hence can be used to score any prospective pose. Indeed, the ShapeFit method was explicitly designed to maximize this probability.
POSIT probabilities were generated using a large testset containing over 25,000 pose predictions and verified through a smaller number (around 100) of predictions that were then validated with X-Ray crystallography. It is important to note that POSIT does not give a probability of binding. Rather it gives a probability that if the ligand does actually bind, what is the likelihood of the POSIT pose being the actual pose.
ShapeFit Method¶
During a drug discovery campaign, thousands of small molecule inhibitors are made in the course of optimizing molecular properties. For projects that have X-Ray crystallographic (XRC) coordinates, structure-based designs help guide the medicinal chemistry efforts. In many cases XRC provides a detailed picture of the binding of a small-molecule inhibitor into the binding site.
Many techniques exist for pose-prediction and are well documented [Erickson-2004] . However, very few provide a probability that the generated pose is correct where correct is typically considered to be less than 2.0 Ångströms RMSD (root mean square distance) from experimental crystal structure. In fact, many docking scores such as Chemscore, Chemgauss3, PLP [Tuccinardi-2010] are not very correlated with correct ligand pose, and worse are not transferable between systems. The best docking score in one system may not even be close to the best docking score in another.
Two definitions will be used during the remainder of this discussion:
Bound-Ligand This is a known, experimentally derived bound ligand from the same protein context in which SHAPEFIT is attempting to find poses of ligands.
Fit-Ligand This is the unknown ligand that is being pose-predicted.
Using the Bound Ligand¶
SHAPEFIT overcomes these issues by comparing predicted poses to observed bound ligands in related co-crystals. As the observed ligand becomes more similar to the the predicted pose, both the binding mode and the shape of the receptor pocket itself tends to become more similar.
The similarity measures being used are 2D path-based fingerprints and the 3D TanimotoCombo [Hawkins-2010] that compares shape and the Mills-Dean approximation of electrostatics [MillsDean-1996] . These similarity measures choose the most appropriate system to dock against and provide a prediction of the quality of the result.
The TanimotoCombo measure is agnostic of how the poses in question are generated; it can be used to validate and provide a pose prediction probability regardless of how the pose was generated. In practice, SHAPEFIT exploits the predictive capabilities of the TanimotoCombo measure by using it in an optimization function that drives a flexible fitting routine.
Essentially, during pose optimization, SHAPEFIT attempts to force a predicted pose into the binding mode of a known ligand. If the induced strain becomes too large, the optimization stops. This final pose is used to predict the overall quality of fit. In this fashion, SHAPEFIT is able to rescue 10-20% of the original rigidly overlaid poses and place them within 2.0 Ångströms RMSD of the experimental crystal structure.
Typically during docking, only the protein structure is used to model unknown structures. Given a molecule that is known to bind, SHAPEFIT searches through XRC coordinates of known ligand-protein complexes, determines the complex best able to predict the pose of the molecule and then generates both a pose and the probability that the pose is correct, usually in well under a minute per ligand.
SHAPEFIT’s basic algorithm:
Given a set of potential complexes, SHAPEFIT chooses the appropriate complex based on the 2D or 3D similarity to the bound ligand. The best complex, in general, has the highest 2D or 3D similarity of the input molecule to the chosen complex’s bound ligand.
After the complex is chosen, a flexible fit is performed that attempts to match the binding mode of the bound ligand using an adiabatic optimization method [Wlodek-2006] . This optimization method is known as the SHAPEFIT potential.
The term adiabatic comes from the Greek “impassable”, and in this case SHAPEFIT sets up a chemical strain boundary that the optimization cannot broach.
SHAPEFIT seeds the flexible fit by expanding the poses generated by the original 3D similarity as described in (1) and then applying the shape constraint of the bound ligand.
As shown in figure *SHAPEFIT* Optimization, SHAPEFIT works by first using the known bound ligand to position the input molecule and follows up by using the bound ligand as a shape constraint during MMFF optimization [Halgren-I-1996] [Halgren-II-1996] [Halgren-III-1996] [Halgren-IV-1996] [Halgren-V-1996] [Halgren-VI-1999] [Halgren-VII-1999] . While the input molecule is being forced into the shape constraint, MMFF strain is monitored to form the adiabatic boundary. When the strain becomes too large, the optimization is reversed or stops altogether.
The interactions from the bound ligand are then used as a further constraint during ligand-protein optimization. This helps to remove clashes with the protein and provide better interactions between un-constrained ligand atoms.
This is a long winded way of saying that SHAPEFIT’s optimization attempts to force the molecule into the known binding mode without creating undue strain on the molecule being placed into the protein.

SHAPEFIT Optimization: Starting from an initial alignment, use the shape constraint of the bound-ligand to drive a flexible fit while simultaneously limiting strain¶
As shown in figure SHAPEFIT Cross Docking Results, analyzing the Kinase data set used in [Tuccinardi-2010] pose-prediction using SHAPEFIT is seen to perform remarkably better for similar ligands than standard docking techniques at higher TanimotoCombo values:
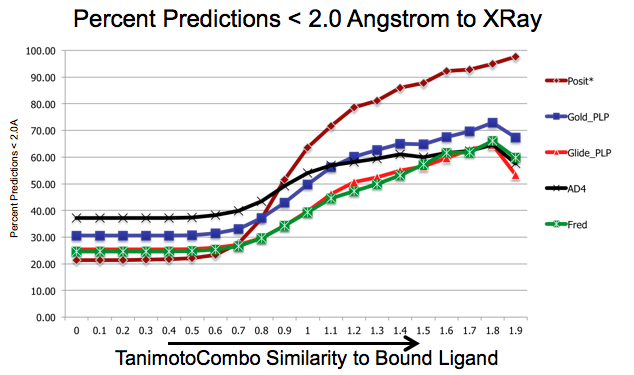
SHAPEFIT Cross Docking Results: Probability of finding a good pose based on bound-ligand fit-ligand TanimotoCombo similarity. Standard docking results are essentially the same and follow the same trajectories flattening out as they hit their limit of accuracy. While SHAPEFIT performs worse at low similarities it continually increases as similarity increases.¶
While SHAPEFIT is not a good technique for determining the pose between known bound-ligands and fit ligands with low similarity, as the similarity increases, the probability of determining the correct pose increases rapidly. The similarity where this crosses over is remarkably small, only around 0.9 TanimotoCombo. This is most likely because as the similarity increases, the active site similarity also increases. Below 0.9 TanimotoCombo POSIT reverts to the Hybrid method.
On Clashes¶
Unlike the FRED and HYBRID methods, SHAPEFIT is heavily biased towards the known bound ligand. In some cases this causes the pose to clash with the protein. This is especially true if the original bound ligand already clashes with the protein.
Currently, we do not filter out clashing poses although options and functionality for controlling this with SHAPEFIT will be investigated in future releases.
Using Multiple Receptors¶
Using multiple receptors to dock or pose molecules has been shown to greatly increase the reliability of docking.
The OEDocking TK can be used to work with multiple receptors by
calling the AddReceptor
with each of the receptors as shown in the following example,
Listing 5. The OEPosit
will automatically detect the best receptor for the ligand in
action and dock to it.
One of the difficulties of docking to multiple receptors, is that
they can consume quite a large amount of memory so holding multiple
docking objects at the same time can be problematic. This can be controlled
with the SetPoserCacheCount
method, based on the available hardware capacity and the size of the
receptors in use.
Receptor Flexibility¶
Pose generated from Posit with the default options can sometimes contain
clashes. Users have the option to perform pose relexation, either on the
clashed poses or on all generated poses, with the appropriate option using the
SetPoseRelaxMode
method.
Note
Turing on receptor flexibility by setting the allowed clash type to anything
other than OEAllowedClashType.ANY can make the
OEPosit calculations extremely time consuming.