OEToolkits 2019.Oct
Spruce TK: A new toolkit for biomolecular structure preparation
The 2019.Oct release introduces Spruce TK. Spruce TK takes experimental biomolecular structure data in either PDB or mmCIF formats, and prepares the structures for use with downstream modeling applications, such as docking with FRED or HYBRID, pose prediction with POSIT, identifying favorable and unfavorable waters with SZMAP or free energy calculations using molecular dynamics simulations.
With Spruce TK, users can easily prepare proteins using either C++ or
Python APIs (see OEMakeDesignUnits), where the output of the file is the recently
developed OEDesignUnit class and is serialized to a new OEDU file format.
The prepared system is componentized into molecular categories including protein, ligand,
nucleic, solvent, and many other molecular types common to biomolecular experiments.
By default, hydrogens are added to systems using a tautomer search for any ligand, cofactor,
or other nonstandard residue molecules. In addition, the system is split into distinct biological
units, and all final design unit structures are superposed into the common frame of reference of a
parent structure. For more details, see Spruce theory.
Spruce TK has been tested across a broad range of industry-relevant targets. It has been used in virtual screening and molecular dynamics simulations in Orion, as well as to prepare structures for loading into the MacroMolecular Data Service (MMDS).
OEChem TK: Fast Substructure Search
Substructure search is a fundamental task in cheminformatics. OEChem TK now provides improved substructure search capability, allowing users to search tens of millions of molecules in seconds.
OEChem TK uses the screen-based approach to accelerate the search process.
Screens are bit vectors encoding global and local features of molecules. They are
prebuilt and can be used to rapidly eliminate structures that could not be
matched to a query molecule. Only molecules that pass the screen are subjected
to the computationally more expensive atom-by-atom matching for validation.
OEChem TK provides three types of built-in screens based on the source of the
query molecule: MDL,
SMARTS, and
Molecule.
All screens are rigorously tested using diverse sets of queries and multiple
molecule databases to ensure that they do not eliminate true positive matches.
The new multi-threaded substructure search can be performed in two modes:
in-memory
and molecule-database.
The in-memory
mode provides the fastest way to search a dataset, but it is memory-intensive
as it holds both screens and molecules in memory.
The molecule-database mode
keeps only the screens in memory; only molecules with unmatching screens have to
be loaded into the memory. This method can be slower but uses significantly
less memory, allowing users to search larger datasets.
screen generation performance |
substructure search performance |
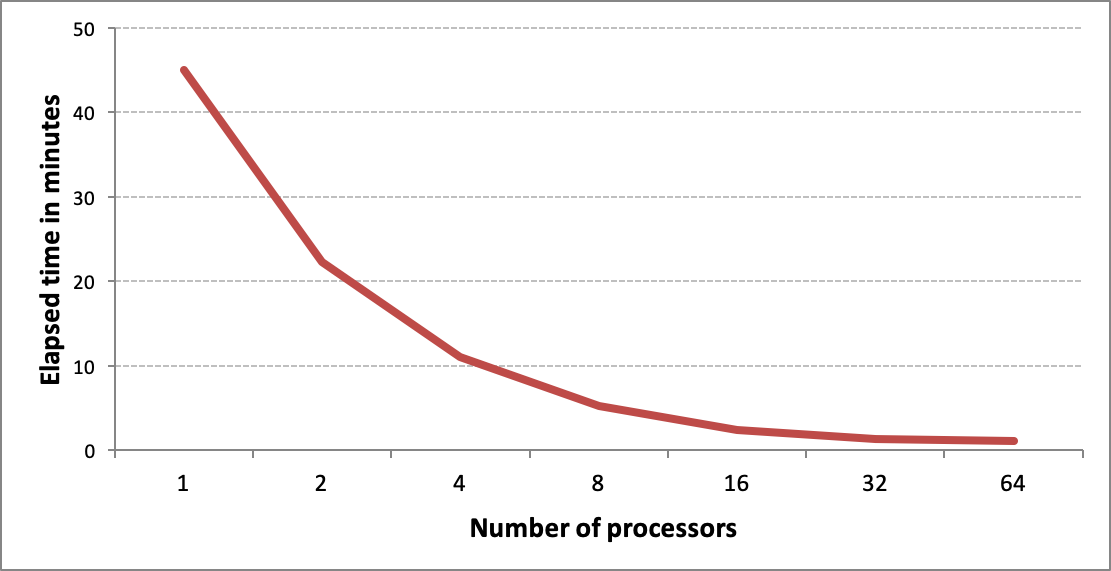
|
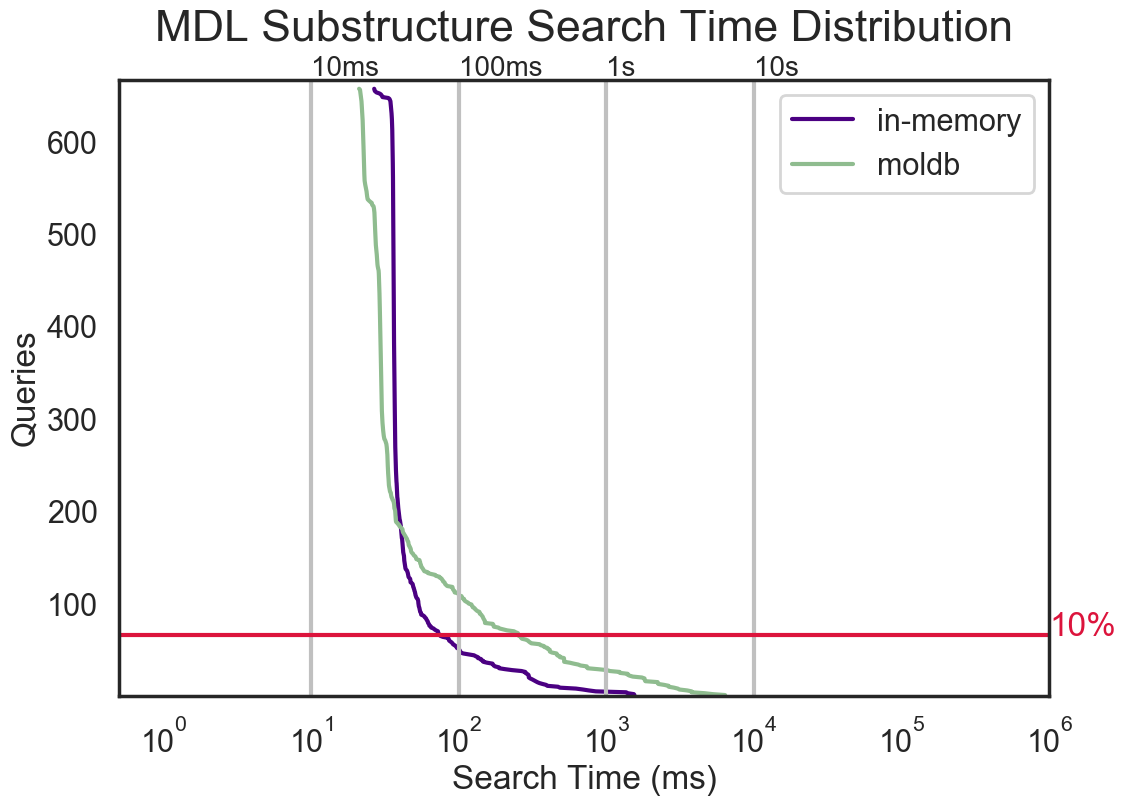
|
See the Molecule Searching chapter of OEChem TK for examples using the new OESubSearchDatabase class. For the full list of new preliminary APIs, see OEChem TK 2.3.0.
OEFF TK: SMIRNOFF
The 2019.Oct release incorporates the SMIRNOFF small molecule force field from the Open Force Field Initiative into OEFF TK. The force field can handle almost all pharmaceutically relevant chemical space.
The SMIRNOFF force field differs from traditional force fields principally in the data representation of the chemistry. It uses extended SMARTS strings for parameters instead of atom types, greatly reducing the number of interdependent parameters. It currently adopts the functional form of the AMBER force field, but the Open Force Field Initiative is actively examining improvements to this. OEFF TK intends to include the most up-to-date version of the force field in its own releases whenever possible.
General Notices
This is the last release to support macOS 10.12. Full support for macOS 10.15 will be added in the next release.
This is the last release to support RHEL6. Full support for RHEL8 will be added in the next release.
The 2019.Oct release skips Java support for macOS. For Java support on RHEL6, please contact support@eyesopen.com.
A new search engine that provides better hits and shows results in a much cleaner format has been added to the documents.
The 2019.Oct release now uses SWIG 3.0.12 for generating C#, Java, and Python wrappers. C# and Python use SWIG’s built-in support for wrapping C++ std::vector and string types.