OEToolkits 2026.1
Release Highlights 2026.1
OEChem TK: Peptide Informatics Support
This release delivers a comprehensive set of peptide informatics capabilities in OEChem TK, including reliable conversion between molecular structures and HELM representations. Support for custom monomer dictionaries enables seamless incorporation of proprietary nonstandard amino acids into peptide discovery workflows. The release also includes a built-in monomer dictionary containing more than 260 monomers commonly encountered in peptide chemistry projects.
In addition, enhanced visualization capabilities in Grapheme TK enable clear and intuitive representation of complex peptides, facilitating the interpretation and communication of diverse peptide chemistries.
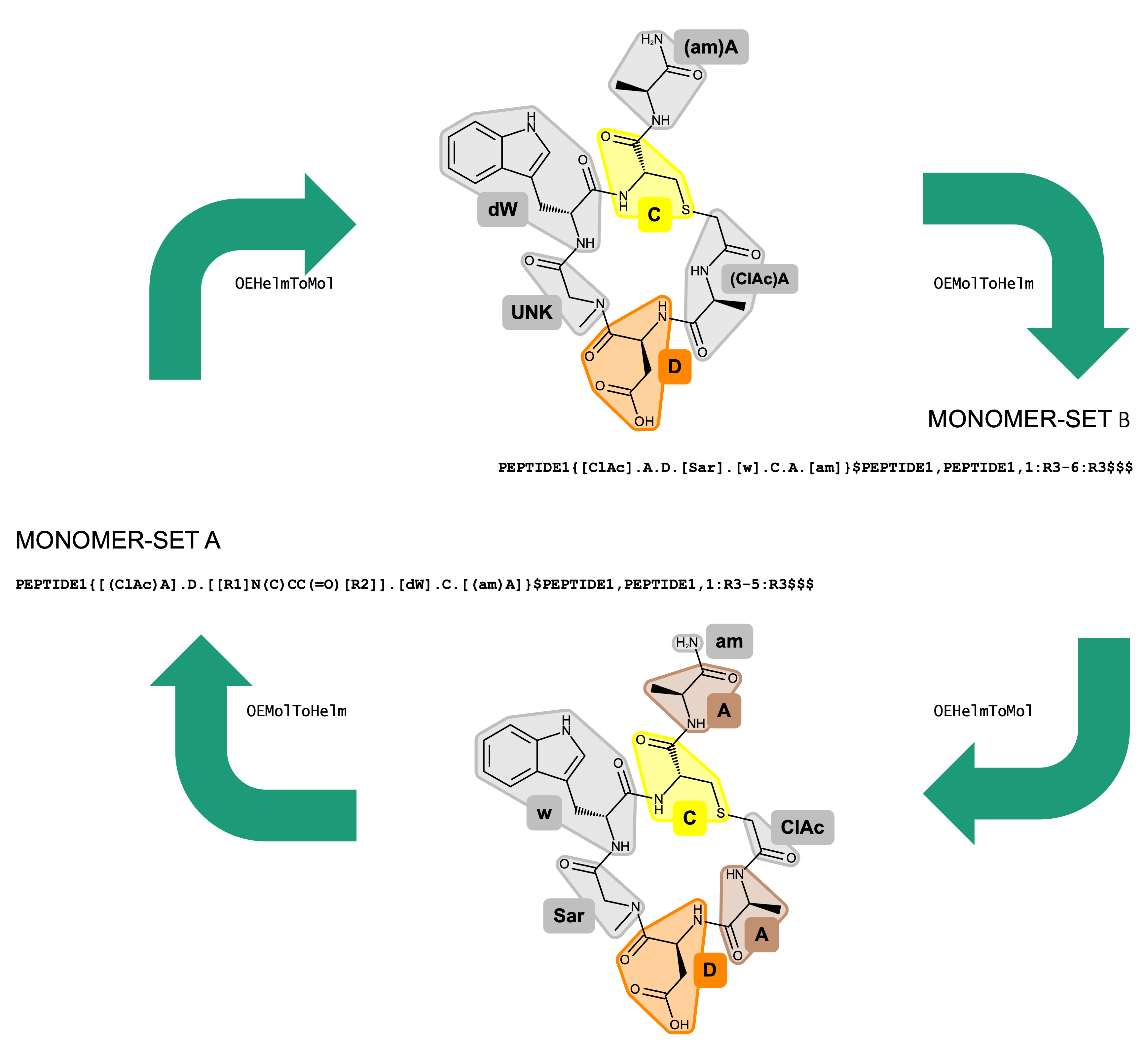
Figure 1. Conversion between HELM strings generated with different monomer sets.
OMEGA: Torsion Driving with Thompson Sampling
Conformer generation using OMEGA with torsion driving now automatically
uses Thompson sampling. The algorithm for Thompson sampling in OMEGA has been
optimized for performance and extended to the generation of larger ensembles, such
as those used in the classic and pose modes of OMEGA.
Thompson sampling is a Bayesian approach that drives conformer generation toward
low energy structures, enabling conformer generation at speed without sacrificing accuracy.
In general, the speed increase with Thompson sampling is larger when working with larger
molecules and generating smaller ensembles. On a dataset of ~40K compounds, compiled
from multiple vendor databases consisting of molecules of varied sizes, an overall
2x speedup over torsion driving without Thompson sampling is observed when generating
conformers in the classic mode. Conformer generation for the same dataset using
the fastrocs mode provides a 3x speedup over torsion driving without Thompson sampling.
A comparison of timings for the dataset of the ~40K compounds from multiple vendor
databases is shown in Figure 2.
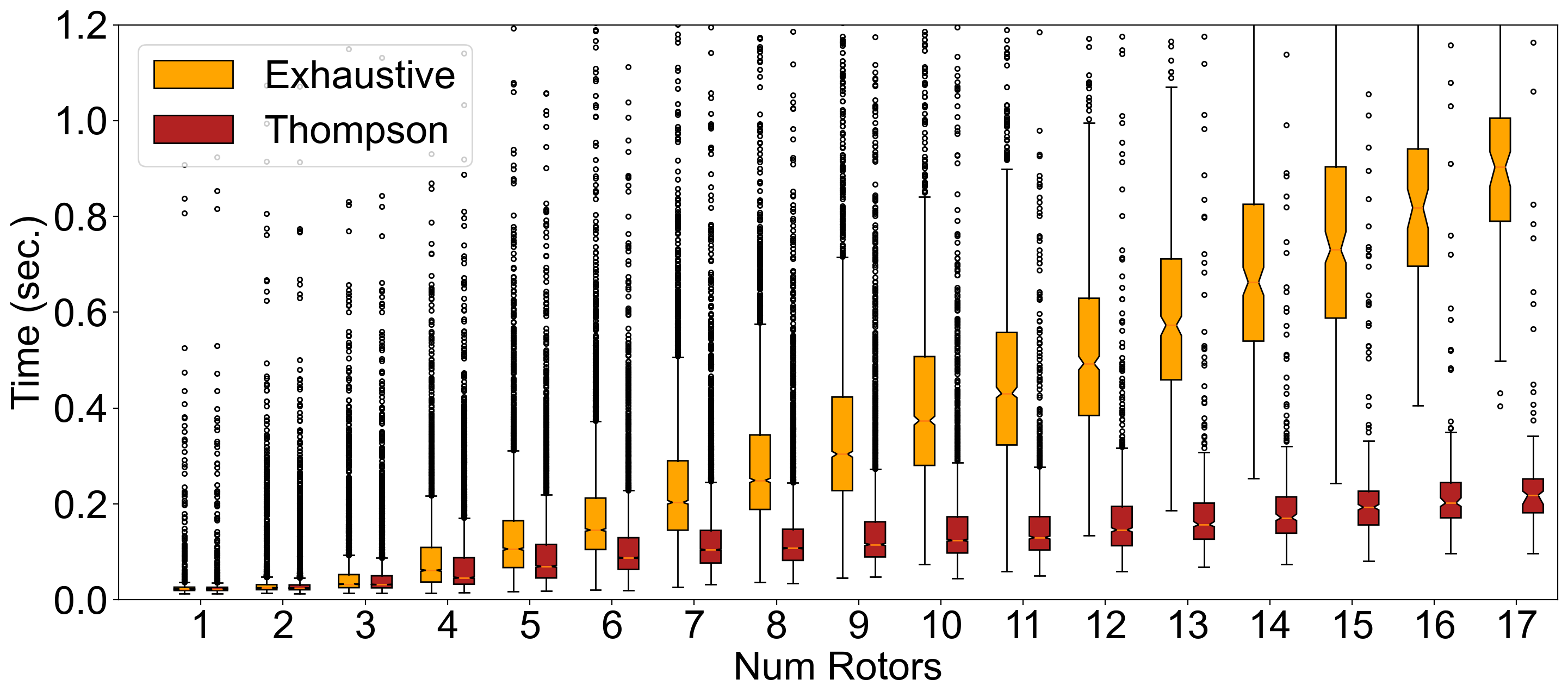
Figure 2. Comparison of OMEGA performance in the classic mode
between exhaustive and Thompson sampling. The box plots show the distribution
of processing time binned by rotor count.
OMEGA maintains high accuracy while achieving this performance gain.
As seen in Figure 3, both exhaustive and Thompson sampling methods in the
classic mode reproduce conformations within 1 Å RMSD of the experimental
conformation for 80% of ligands across the 6,000 PDB structures with HT
(Highly Trustworthy) Iridium scores. Structure-based virtual
screening performance is equivalent to that obtained from exhaustive sampling.
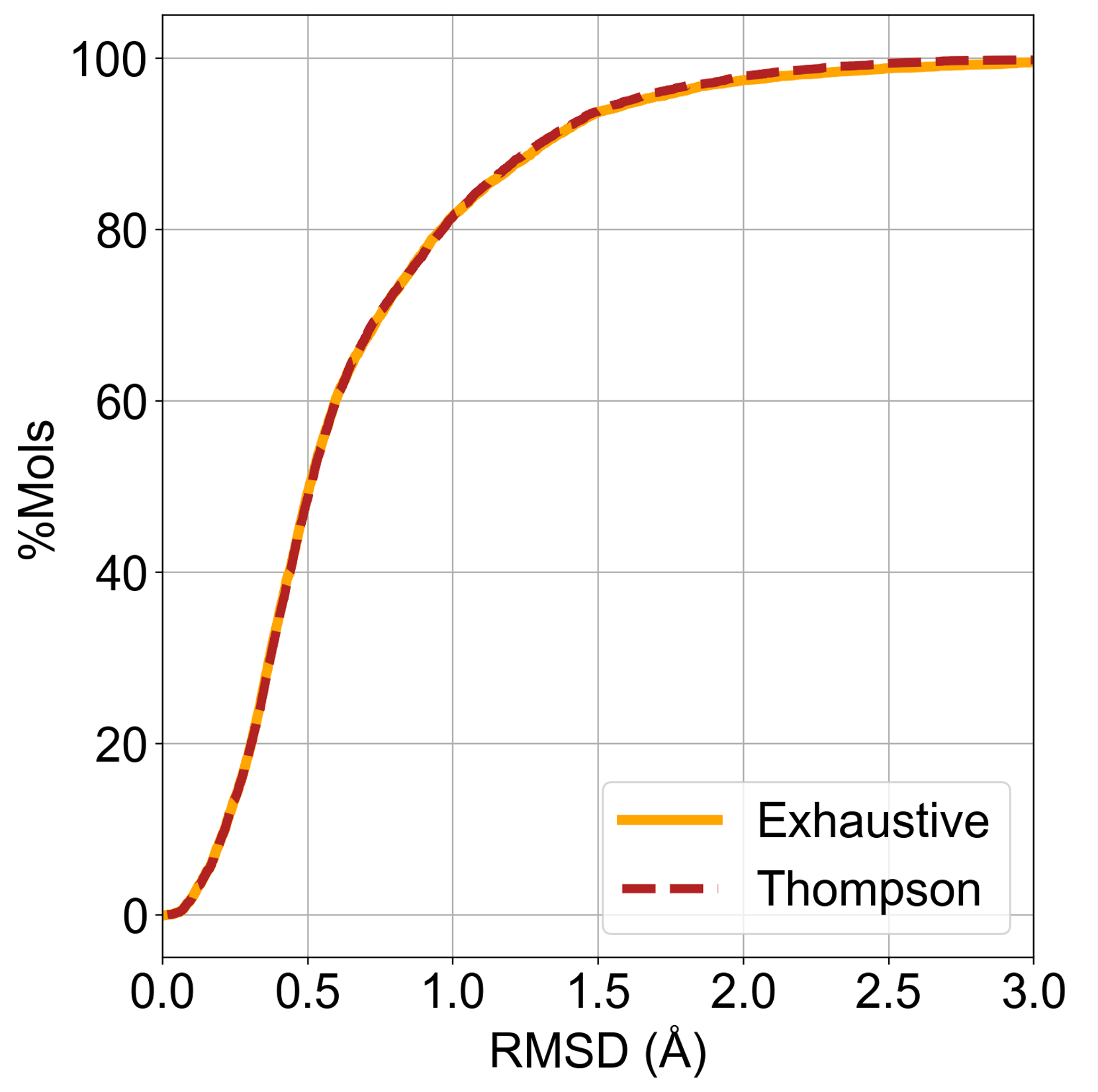
Figure 3. Distribution of RMSD between the best pose from the generated ensemble
and the crystal pose for the two sampling methods using classic mode in OMEGA.
Bioisostere TK: Multiquery Replacements
Bioisostere TK now includes new API functionality for simultaneous replacement of multiple query fragments in a molecule. With the addition of the new functionality, Bioisostere TK is uniquely positioned with tools to build applications for 3D generative de novo molecule design.
In addition to the ability to simultaneously replace multiple query fragments, tools have been added to obtain scores of multiple replacements from their single replacement counterparts, building a single hit list of Brood overlay scores obtained from multiple query replacements, building hit lists combining hits from different queries and multiple queries, and building clusters from the combined hit lists. All of the API designs are compatible for use with an arbitrary number of queries on a molecule, without any limitations. With the addition of each query, the searchable space can be expanded combinatorially, enabling exploration of increasingly larger chemical design spaces.
As shown in Figure 4, users can select specific fragments in a molecule of interest and generate targeted replacement suggestions.
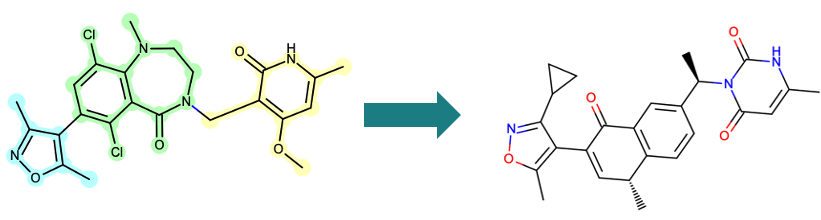
Figure 4. De novo molecule design of an EZH2 inhibitor. The compound on the left highlights the input molecule being divided into three fragments to build three separate Brood queries. The compound on the right is a new molecule designed using Bioisostere TK to replace all three queries from the left.
Supported Platforms
Package
Versions
Linux
Windows
macOS
Python
3.11 - 3.14
RHEL8/9/10, Ubuntu22/22-ARM/24
Win11
14, 15, 26
C++
RHEL8/9/10, Ubuntu22/22-ARM/24
Win11 (VS2022)
14, 15, 26
Java
8, 11, 21
RHEL8/9/10, Ubuntu22/22-ARM/24
Win11
14, 15, 26
C#
Win11 (VS2022)
General Notices
This is the last release of the OpenEye Java and C# toolkits. Existing versions, including the current release versions, will continue to be available.
Python Support:
OpenEye Python Cookbook is now available as a pip-installable package.
Support for Python 3.14 has been added in this release for all supported platforms.
Support for Python 3.10 has been dropped in this release.
Support for Python 3.11 will be dropped in the winter of 2027.
Linux Support:
Support for Red Hat 10 has been added in this release.
Support for Ubuntu 26 will be added in the winter of 2027.
Support for Ubuntu 22 will be dropped in the winter of 2027.
macOS Support:
Support for macOS 26 has been added in this release.
Support for macOS 13 has been dropped.
Support for all Intel-based Macs will be dropped in the winter of 2027.
Support for Windows 10 has been dropped in this release.