ShapeFit
SHAPEFIT is a pose prediction method that is built up on the concept that similar ligands have similar binding modes in a protein active site. Given a molecule that is known to bind, and a ligand-protein complex containing a similar ligand or the binding pose of a similar ligand, SHAPEFIT overlays the docked ligand onto the known bound ligand. The approach used here is an extension of the molecular- shape-based ligand-alignment algorithm used in ROCS. Although both are shape-based ligand alignment, unlike ROCS, SHAPEFIT allows for for intra-molecular flexibility of the docked ligand. SHAPEFIT also guards against the docked ligand bumping into the protein, when using a ligand-protein complex as reference system.
When multiple receptors, or bound ligand references are provided, SHAPEFIT searches through XRC coordinates of ligands, determines the bound-liagnd best able to predict the pose of the molecule and then generates both a pose and the probability that the pose is correct.
SHAPEFIT’s basic algorithm:
Given a set of potential complexes or bound-ligands, SHAPEFIT chooses the appropriate reference system based on the 2D/3D similarity to the bound ligand. The best reference, in general, has the highest 2D/3D similarity of the input molecule to the chosen bound ligand.
After the complex is chosen, a flexible fitting is performed that attempts to maximize the shape and color similarities between the input molecule and the bound ligand while at the same time minimizing the intra-molecular force field on input molecule.
SHAPEFIT seeds the flexible fit by expanding the poses generated by the original 3D similarity as described in (1) and then applying the shape constraint of the bound ligand.
As shown in figure SHAPEFIT Optimization, SHAPEFIT works by first using the known bound ligand to position the input molecule and follows up by using the bound ligand as a shape constraint during force field optimization [Halgren-I-1996] [Halgren-II-1996] [Halgren-III-1996] [Halgren-IV-1996] [Halgren-V-1996] [Halgren-VI-1999] [Halgren-VII-1999] . While the input molecule is being forced into the shape constraint, the generated pose strain is monitored to avoid generating high strain poses.
The interactions between the generated poses and proteins are utilized to identify clash-free poses while simultaneously selecting the poses with the highest Pose probability.
This is a long winded way of saying that SHAPEFIT’s optimization attempts to force the molecule into the known binding mode without creating undue strain on the molecule being placed into the protein.
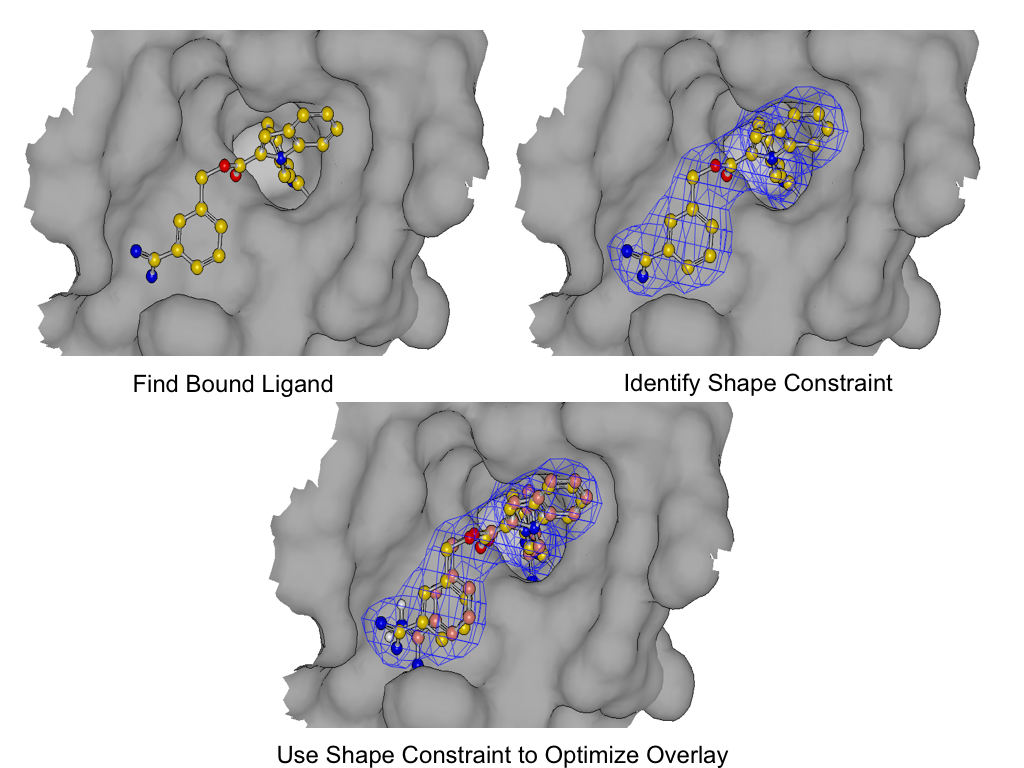
SHAPEFIT Optimization: Starting from an initial alignment, use the shape constraint of the bound-ligand to drive a flexible fit while simultaneously minimizing intra-molecular force field of the fit molecule
As shown in figure SHAPEFIT Cross Docking Results, analyzing the Kinase data set used in [Tuccinardi-2010] pose-prediction using SHAPEFIT is seen to perform remarkably better for similar ligands than standard docking techniques at higher TanimotoCombo values:
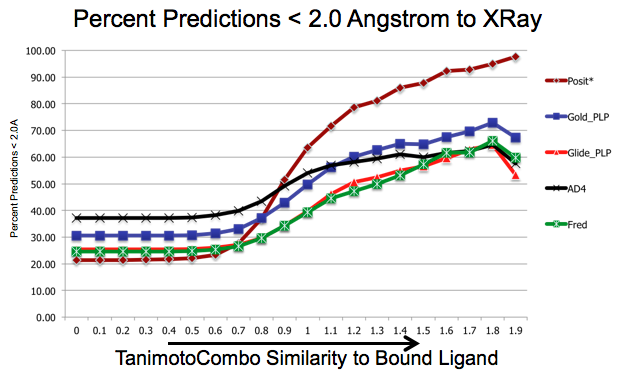
SHAPEFIT Cross Docking Results: Probability of finding a good pose based on bound-ligand fit-ligand TanimotoCombo similarity. Standard docking results are essentially the same and follow the same trajectories flattening out as they hit their limit of accuracy. While SHAPEFIT performs worse at low similarities it continually increases as similarity increases.
While SHAPEFIT is not a good technique for determining the pose between known bound-ligands and fit ligands with low similarity, as the similarity increases, the probability of determining the correct pose increases rapidly. This is most likely because as the similarity increases, the active site similarity also increases.
On Clashes
Unlike the FRED and HYBRID methods, SHAPEFIT is heavily biased towards the known bound ligand. In some cases this causes the pose to clash with the protein. This is especially true if the original bound ligand already clashes with the protein.