Matched Pair analysis using a multi-process generation of an MMP index
A program that performs a multi-process matched pair analysis of a set of structures for indexing and saves the generated index file for subsequent loading and querying. This example uses the python multiprocessing module.
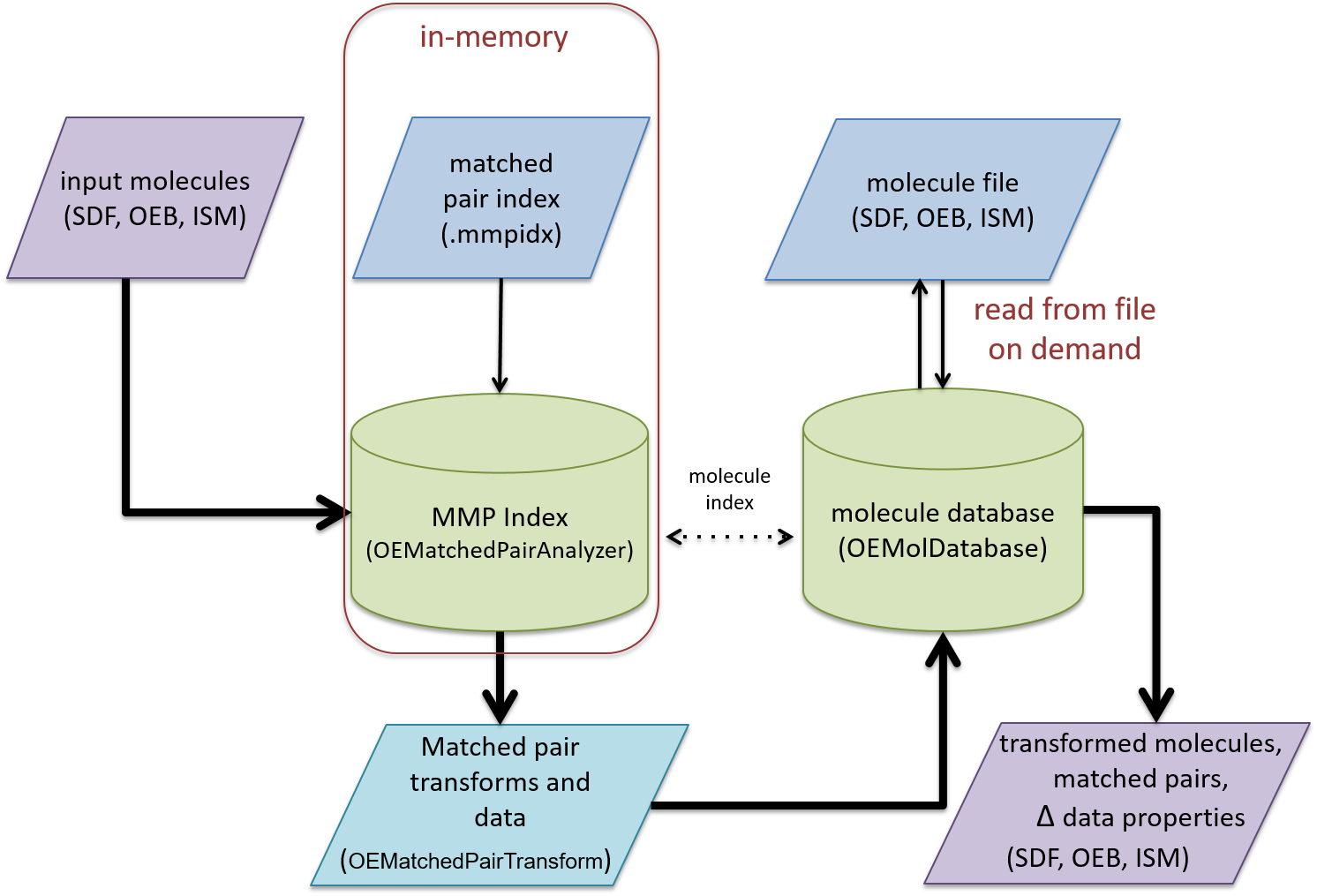
Schematic representation of the Matched Pair Analysis process
See also
OEMatchedPairAnalyzer class
OEMatchedPairApplyTransformsfunction
Command Line Interface
prompt> CreateMMPIndexParallel.py [ -pool num_processes ] index.sdf output.mmpidx
Code
Download code