Protein Preparation
Protein preparation is the process of transforming a macromolecular structure into a form more suitable for a computational experiment. The specific set of tasks required depends on the experiment and the source of the structure. Tools for several common prep tasks are described below. For a demonstration of how these can be combined, see the Preparing a Protein example.
For a more comprehensive protein preparation process, Advanced Macromolecular Preparation Workflow shows an end-to-end preparation workflow to process protein structures for molecular modeling. For more information on Spruce TK see the associated Theory and Spruce API.
Processing Alternate Locations
Protein structures rarely describe whole molecules. Instead, they are usually
missing atoms that could not be resolved well. In addition, many structures
contain multiple copies of atoms called alternate (“alt”) locations
or alternate conformations that can indicate that part of the structure is
moving. As the resolution of X-ray crystal structures improves, alt locations
can be expected to become ubiquitous. These conformations are indexed by an
ID character such as A or B that is stored in the residue information
for that atom (see Biopolymer Residues).
Having extra copies of atoms can interfere with some experiments, such as
docking. One way to handle this is to eliminate all but one conformation.
By default, OEChem TK drops all but the A or blank conformations when
reading atoms in a PDB format file. To prevent this, the file
must be read with the OEIFlavor_PDB_ALTLOC flavor.
Listing 1: Setting the PDB Flavor to Retain All Alts
ims = oechem.oemolistream()
ims.SetFlavor(oechem.OEFormat_PDB, oechem.OEIFlavor_PDB_ALTLOC)
ims.SetFlavor(oechem.OEFormat_MMCIF, oechem.OEIFlavor_MMCIF_ALTLOC)
Another way to drop all but one conformation is to use the OEAltLocationFactory object (see the Alternate Locations section) and generate, for example, a molecule that retains only the highest occupancy conformation of each location group. This object can be used to iterate through each combination of alt locations (for example, when a group is moving in an active site).
Listing 2: Keeping the Highest Occupancy Alt Locations
alf = oechem.OEAltLocationFactory(mol)
if alf.GetGroupCount() != 0:
alf.MakePrimaryAltMol(mol)
Adding Hydrogens and Optimizing Hydrogen Bonds
Traditionally, most protein structures have been generated with either no
explicit hydrogens or only polar hydrogens.
However, having all atoms explicitly represented and positioned to make
appropriate hydrogen bonds is sometimes necessary.
The function OEPlaceHydrogens does this by adding and
placing hydrogens and by “flipping” certain functional groups
(e.g., sidechain amides and imidazoles) as required.
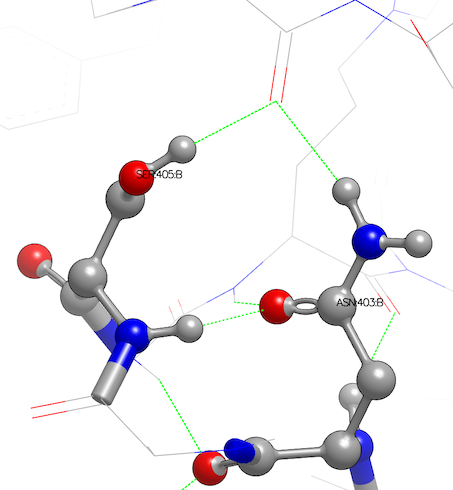
Asparagine has been flipped and serine oriented to H-bond with main-chain atoms
Listing 3: Adding and Optimizing the Positions of Hydrogens
if oechem.OEPlaceHydrogens(mol):
# ...
There are several OEPlaceHydrogensOptions parameters that control the process. For example, it is possible to prevent the bond lengths of any covalent bonds to pre-existing hydrogens from being altered:
Listing 4: Setting Options for Placing Hydrogens
opt = oechem.OEPlaceHydrogensOptions()
opt.SetStandardizeBondLen(False)
There are also OEPlaceHydrogensOptions
parameters for restraining which states a mover group is sampled over.
Listing 5 shows a predicate for identifying
atoms belonging to a specific residue.
Listing 5: Predicate for Residue Equivalence
class IsSameResidue(oechem.OEUnaryAtomPred):
def __init__(self, res):
oechem.OEUnaryAtomPred.__init__(self)
self.referenceResidue = res
def __call__(self, atom):
res = oechem.OEAtomGetResidue(atom)
return oechem.OESameResidue(res, self.referenceResidue)
def CreateCopy(self):
# __disown__ is required to allow C++ to take ownership of this
# object and its memory
return IsSameResidue(self.referenceResidue).__disown__()
We can use that predicate to prevent a selected residue from flipping:
Listing 6: Preventing a Residue from Flipping
# given predicate IsSameResidue, residue selectedResidue and molecule mol...
opt = oechem.OEPlaceHydrogensOptions()
opt.SetNoFlipPredicate(IsSameResidue(selectedResidue))
if oechem.OEPlaceHydrogens(mol, opt):
# selectedResidue will not be flipped...
Using an OEPlaceHydrogensDetails object enables examination of the results, showing the clusters of movable groups explored and the scores. A simple way to get at this information is to display a summary description:
Listing 7: Examining the Details of Placing Hydrogens
# given molecule mol and OEPlaceHydrogensOptions opt...
details = oechem.OEPlaceHydrogensDetails()
if oechem.OEPlaceHydrogens(mol, details, opt):
print(details.Describe())
The following is a fragment from a details object description:
OEBio 2.1.1: score system MMFF-NIE: flip bias 2.000
87 clusters : 5 flips
cluster 0 : score -53.935!
CG ASN44(A) : amide: -6.084 (o=-53.93!,f=-8.42!)
NZ LYS47(A) : NH3 150deg: -14.865
CG HIS80(A) : +bothHN: -5.628 (o=-53.93!,f=-15.89!)
OG1 THR81(A) : OH 65deg: 1.255
CD GLN87(A) :FLIP amide: -4.976 (o=-26.48!,f=-53.93!)
CG ASN126(A) : amide: -5.193 (o=-53.93!,f=-35.33!)
CG ASN169(A) : amide: -2.230 (o=-53.93!,f=-33.02!)
CD GLN203(A) : amide: -1.400 (o=-53.93!,f=-15.53!)
CG HIS205(A) : +bothHN: -8.693! (o=-53.93!,f=-6.89!)
OG1 THR232(A) : OH 179deg: 9.120
O3B XYP601(A) : OH 280deg: -5.020
O2B XYP601(A) : OH 173deg: 3.917
O4B XYP601(A) : OH 50deg: 1.236
O3 XIF602(A) : OH 29deg: -3.688
cluster 1 : score -85.724!
CG HIS85(A) : +bothHN: -7.737! (o=-85.72!,f=-84.85!)
OG SER86(A) : OH 241deg: 2.116
OG SER139(A) : OH 185deg: 3.367!
OH TYR171(A) : OH 25deg: -3.384
CG ASN172(A) : amide: 1.153 (o=-85.72!,f=-68.75!)
NZ LYS179(A) : NH3 188deg: -39.497
...
single 13 : OG1 THR2(A) : OH 151deg: 1.545
single 14 : OG SER25(A) : OH 292deg: 2.046
single 15 : OH TYR29(A) : OH 199deg: -1.262
single 16 : OG SER35(A) : OH 254deg: 1.935
...
See also
the
OEPlaceHydrogensDetails.Describemethod for more details about the output above and theMMFF-NIEscoring system that generated it.
Splitting Macromolecular Complexes
One common step in preparing a macromolecular complex for use as input to a calculation is to separate components based on their functional roles. For example, a SZMAP calculation on a four-chain protein crystal structure might require one of four copies of the ligand to be extracted and the protein-complex associated with that ligand binding site to be separately extracted, ignoring any waters, buffers, etc.
Listing 8 uses the
OESplitMolComplex function to separate the input molecule
into the ligand for the first binding site, the protein-complex
(including cofactors) for that site, the binding site waters,
and everything else. In this example, the ligand is identified automatically
and the binding site contains components near the ligand. If a ligand cannot be
located, the entirety of each protein and everything nearby defines each
binding site. If the input molecule is empty or there is some other
problem that prevents processing, OESplitMolComplex
will return false.
Listing 8: Splitting an Input Molecule
# for input molecule inmol ...
lig = oechem.OEGraphMol()
prot = oechem.OEGraphMol()
wat = oechem.OEGraphMol()
other = oechem.OEGraphMol()
if oechem.OESplitMolComplex(lig, prot, wat, other, inmol):
# work with the output molecules lig, prot, ...
Listing 8 uses the defaults,
so no attempt would be made to recognize covalent ligands.
Listing 9 uses an option
to turn on the splitting of covalent ligands. There are a
large number of options to control the process of splitting
a molecule. See OESplitMolComplexOptions
for a complete list.
Listing 9: Splitting Covalent Ligands
# given input and output molecules ...
opt = oechem.OESplitMolComplexOptions()
opt.SetSplitCovalent()
if oechem.OESplitMolComplex(lig, prot, wat, other, inmol, opt):
# ...
A related approach uses OEGetMolComplexComponents
to return an iterator of molecules.
Listing 10 also illustrates
providing an explicit ligand residue name that must match
for the ligand to be identified.
Listing 10: Iterating Over Split Components
# for input molecule mol and string ligName ...
opt = oechem.OESplitMolComplexOptions(ligName)
for frag in oechem.OEGetMolComplexComponents(mol, opt):
# ...
The process used by the OESplitMolComplex
and OEGetMolComplexComponents functions involves
two steps:
separating components and assigning functional roles
filtering by roles to select the desired components
OESplitMolComplex accesses separate filters
for lig, prot, wat, and other from the
OESplitMolComplexOptions object.
In OEGetMolComplexComponents,
the combination of the ligand, protein, and water filters
is used to select the output components. The options method
OESplitMolComplexOptions.ResetFilters
can be used for basic changes to the filters, such as selecting
a different binding site (see Listing 11).
Listing 11: Resetting Filters for All Sites
opt = oechem.OESplitMolComplexOptions()
allSites = 0
opt.ResetFilters(allSites)
The function OECountMolComplexSites
returns the number of binding sites.
Listing 12
shows a more involved case, combining protein-complex
and binding-site waters into
the protein filter and nothing in the water filter.
Listing 12: Modifying Filters
opt = oechem.OESplitMolComplexOptions()
p = opt.GetProteinFilter()
w = opt.GetWaterFilter()
opt.SetProteinFilter(oechem.OEOrRoleSet(p, w))
opt.SetWaterFilter(oechem.OEMolComplexFilterFactory(oechem.OEMolComplexFilterCategory_Nothing))
Listing 13
illustrates using an explicit filter when
iterating. OEMolComplexFilter objects
can be of arbitrary complexity.
Listing 13: Explicit Filter When Iterating
# for input molecule mol and options opt ...
for lig in oechem.OEGetMolComplexComponents(mol, opt, opt.GetLigandFilter()):
# ...
For more complex workflows, it is useful to explicitly separate
the tasks of assigning roles and filtering, if only to avoid
the expense of assigning roles multiple times.
Listing 14
shows how to assign roles to each fragment in a molecule
for later use.
Listing 14: Assigning Roles to Each Mol Fragment
# for input molecule mol and options opt ...
frags = oechem.OEAtomBondSetVector()
if oechem.OEGetMolComplexFragments(frags, mol, opt):
# ...
In Listing 15,
the role information for each fragment is used to determine the
number of binding sites and is filtered
to produce ligand and protein complex mols.
Listing 15: Filtering Mol Fragments
# for options opt and OEAtomBondSetVector frags produced earlier ...
numSites = oechem.OECountMolComplexSites(frags)
oechem.OEThrow.Verbose("sites %d" % numSites)
lig = oechem.OEGraphMol()
ligfilter = opt.GetLigandFilter()
if not oechem.OECombineMolComplexFragments(lig, frags, opt, ligfilter):
oechem.OEThrow.Warning("Unable to combine ligand frags from %s" % mol.GetTitle())
protComplex = oechem.OEGraphMol()
p = opt.GetProteinFilter()
w = opt.GetWaterFilter()
if not oechem.OECombineMolComplexFragments(protComplex, frags, opt, oechem.OEOrRoleSet(p, w)):
oechem.OEThrow.Warning("Unable to combine complex frags from %s" % mol.GetTitle())
The basic idea behind automatic ligand identification is
that the list of compounds that are not ligands (cofactors,
buffers, etc.) grows faster than the list of ligands.
The OEResidueCategoryData object
contained within an OESplitMolComplexOptions
is a database of these non-ligands. Although this database
is being updated as we run across more cofactors, buffers, etc.,
it is possible for users to update the database as needed.
Listing 16 shows how
this is done, involving the Russian nesting doll-like organization of a
OEResidueCategoryData within a
OEMolComplexCategorizer within a
OESplitMolComplexOptions object.
Listing 16: Adding a Cofactor
db = oechem.OEResidueCategoryData()
db.AddToDB(oechem.OEResidueDatabaseCategory_Cofactor, "MTQ")
cat = oechem.OEMolComplexCategorizer()
cat.SetResidueCategoryData(db)
opt = oechem.OESplitMolComplexOptions()
opt.SetCategorizer(cat)
Note
Please help OpenEye maintain and expand the built-in OEResidueCategoryData by informing us of any oversights or mistakes.
The most important OESplitMolComplexOptions parameters
can be controlled from the command line using
OEConfigureSplitMolComplexOptions and
OESplitMolComplexSetup
(see Listing 17).
Listing 17: Command Line Options
import sys
from openeye import oechem
def main(argv=[__name__]):
itf = oechem.OEInterface(InterfaceData)
oechem.OEConfigureSplitMolComplexOptions(itf)
if not oechem.OEParseCommandLine(itf, argv):
oechem.OEThrow.Fatal("Unable to interpret command line!")
opts = oechem.OESplitMolComplexOptions()
oechem.OESetupSplitMolComplexOptions(opts, itf)