Working with Szmap TK
The Szmap TK has a simple API, consisting of a few basic API and a couple of objects to configure the calculations (OESzmapEngine and OESzmapEngineOptions) and hold calculated results (OESzmapResults).
Preparing Protein and Ligand Molecules
To perform the necessary electrostatics calculations,
a protein or any other molecular context
being analyzed with SZMAP must already
have all hydrogens explicitly represented as well as have charges and radii assigned
before it can be used. This is done automatically as part of protein preparation
with Spruce TK (see OEMakeDesignUnits).
Charges can also be assigned using Quacpac TK (see OEAssignCharges).
Calculating Energies
To calculate probe energies, we initialize
a OESzmapEngine with a molecular
context, test if the point of interest is clashing,
and if not, calculate the energies. In the example below,
the energies at each ligand atom coordinate
are returned in a OESzmapResults
object, which is accessed for specific values.
Alternatively, OECalcSzmapValue could
be used to get a single OEEnsemble
value directly.
def GetSzmapEnergies(lig, prot):
"""
run szmap at ligand coordinates in the protein context
@rtype : None
@param lig: mol defining coordinates for szmap calcs
@param prot: context mol for szmap calcs (must have charges and radii)
"""
print("num\tatom\t%s\t%s\t%s\t%s\t%s"
% (oeszmap.OEGetEnsembleName(oeszmap.OEEnsemble_NeutralDiffDeltaG),
oeszmap.OEGetEnsembleName(oeszmap.OEEnsemble_PSolv),
oeszmap.OEGetEnsembleName(oeszmap.OEEnsemble_WSolv),
oeszmap.OEGetEnsembleName(oeszmap.OEEnsemble_VDW),
oeszmap.OEGetEnsembleName(oeszmap.OEEnsemble_OrderParam)))
coord = oechem.OEFloatArray(3)
sz = oeszmap.OESzmapEngine(prot)
rslt = oeszmap.OESzmapResults()
for i, atom in enumerate(lig.GetAtoms()):
lig.GetCoords(atom, coord)
if not oeszmap.OEIsClashing(sz, coord):
oeszmap.OECalcSzmapResults(rslt, sz, coord)
print("%2d\t%s\t%.3f\t%.3f\t%.3f\t%.3f\t%.3f"
% (i, atom.GetName(),
rslt.GetEnsembleValue(oeszmap.OEEnsemble_NeutralDiffDeltaG),
rslt.GetEnsembleValue(oeszmap.OEEnsemble_PSolv),
rslt.GetEnsembleValue(oeszmap.OEEnsemble_WSolv),
rslt.GetEnsembleValue(oeszmap.OEEnsemble_VDW),
rslt.GetEnsembleValue(oeszmap.OEEnsemble_OrderParam)))
else:
print("%2d\t%s CLASH" % (i, atom.GetName()))
The five OEEnsemble values
in the example above are the primary ones
for understanding solvent/ligand competition.
See also
GetSzmapEnergies example
Generating Probe Orientations
In addition to energy calculations, the Szmap TK
can be used to generate 3D conformations of probe
molecules at calculated points, annotated
with OEComponent
energies for each conformation. In the example below,
individual atoms are also generated at calculation
points, each annotated with OEEnsemble
values.
def GenerateSzmapProbes(oms, cumulativeProb, lig, prot):
"""
generate multiconf probes and data-rich points at ligand coords
@rtype : None
@param oms: output mol stream for points and probes
@param cumulativeProb: cumulative probability for cutoff of point set
@param lig: mol defining coordinates for szmap calcs
@param prot: context mol for szmap calcs (must have charges and radii)
"""
coord = oechem.OEFloatArray(3)
sz = oeszmap.OESzmapEngine(prot)
rslt = oeszmap.OESzmapResults()
points = oechem.OEGraphMol()
points.SetTitle("points %s" % lig.GetTitle())
probes = oechem.OEMol()
for i, atom in enumerate(lig.GetAtoms()):
lig.GetCoords(atom, coord)
if not oeszmap.OEIsClashing(sz, coord):
oeszmap.OECalcSzmapResults(rslt, sz, coord)
rslt.PlaceNewAtom(points)
clear = False
rslt.PlaceProbeSet(probes, cumulativeProb, clear)
oechem.OEWriteMolecule(oms, points)
oechem.OEWriteMolecule(oms, probes)
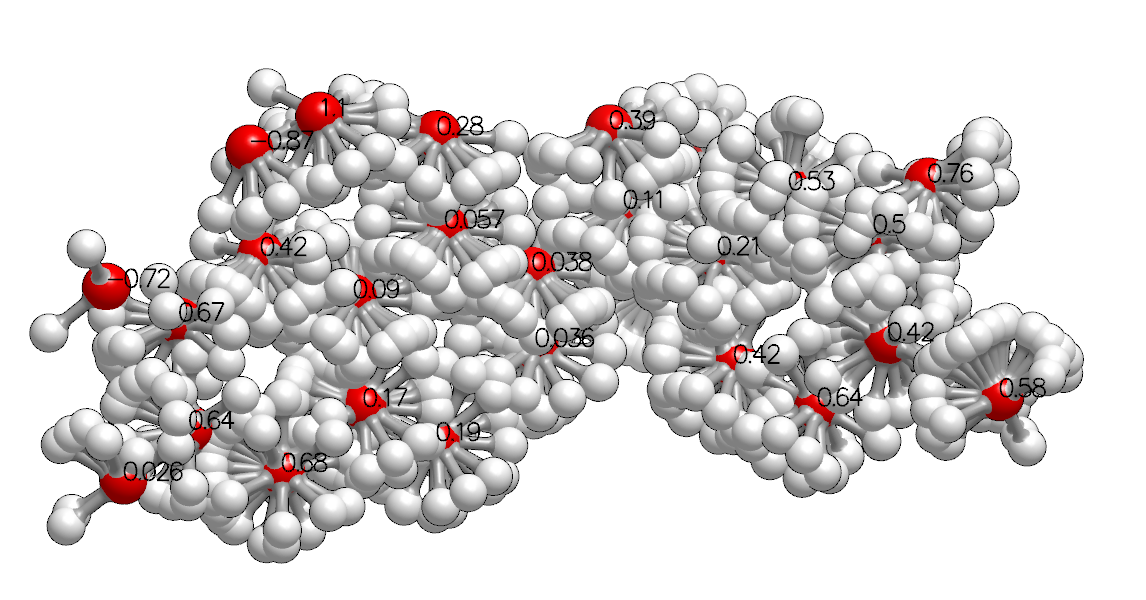
High Probability Probe Orientations and Points with Generic Data Annotation
See also
SzmapBestOrientations example