OESzmapResults
class OESzmapResults
This class represents OESzmapResults, a container for the
results of OESzmap calculations with the
function OECalcSzmapResults.
See also
GetSzmapEnergies example
SzmapBestOrientations example
Constructors
OESzmapResults()
OESzmapResults(const OESzmapResults &rhs)
Default and copy constructors.
Typically an empty, default OESzmapResults
is passed to OECalcSzmapResults
where it is filled with the calculated results.
rslt = oeszmap.OESzmapResults()
operator=
OESzmapResults &operator=(const OESzmapResults &rhs)
Assignment operator.
operator bool
operator bool() const
True indicates that OECalcSzmapResults was called
with a valid OESzmapEngine when this object was created.
Clear
void Clear()
Resets this object to an uninitialized, empty state.
GetComponent
bool GetComponent(double *compArray, unsigned int componentType) const
OESystem::OEIterBase<double> *GetComponent(unsigned int componentType) const
Returns the calculated values of a particular OEComponent,
specified by componentType,
for the 3D point provided to OECalcSzmapResults.
Component values for each probe orientation are the low-level data
used to compose OEEnsemble values
(see OESzmapResults.GetEnsembleValue).
The number of values in the output compArray or the iterator is
OESzmapResults.NumOrientations and values are returned
in the same order as the probe orientations.
coulomb = rslt.GetComponent(oeszmap.OEComponent_Interaction)
print("interaction:")
print(" ".join("%.3f" % c for c in coulomb))
Returns false or an empty iterator if the OEComponent
type is not recognized.
GetCoords
bool GetCoords(float *xyz) const
bool GetCoords(double *xyz) const
Returns the coordinates of the 3D point where calculations were performed by
OECalcSzmapResults to create this object.
The point is passed back in a float or double array of size three with coordinates in {x,y,z} order.
point = oechem.OEFloatArray(3)
rslt.GetCoords(point)
Returns false if this OESzmapResults is uninitialized.
GetEnsembleValue
double GetEnsembleValue(unsigned int ensembleType) const
Returns the calculated value of a particular OEEnsemble,
specified by ensembleType,
for the 3D point provided to OECalcSzmapResults.
Ensemble values are the results of calculations
over all orientations of the probe.
In general, these are built by Boltzmann summation of
various combinations of OEComponent values
(see OESzmapResults.GetComponent).
nddg = rslt.GetEnsembleValue(oeszmap.OEEnsemble_NeutralDiffDeltaG)
Returns 0.0 if the OEEnsemble
type is not recognized or this OESzmapResults is uninitialized.
GetProbabilities
bool GetProbabilities(double *probArray) const
OESystem::OEIterBase<double> *GetProbabilities() const
Returns the statistical mechanical probabilities for each probe orientation
at the 3D point provided to OECalcSzmapResults.
Probability values can be used to Boltzmann weight
OEComponent values and are used to select which
probe orientations are returned by OESzmapResults.PlaceProbeSet.
The number of values in the output probArray or the iterator is
OESzmapResults.NumOrientations and values are returned
in the same order as the probe orientations.
prob = rslt.GetProbabilities()
print("greatest prob = %.3f" % prob[order[0]])
Returns false or an empty iterator if this OESzmapResults is uninitialized.
GetProbabilityOrder
bool GetProbabilityOrder(unsigned int *orderArray) const
OESystem::OEIterBase<unsigned int> *GetProbabilityOrder() const
Returns an array or iterator of indices referring to probe orientations or
associated OEComponent and probability values,
sorted in the order of increasing probability
(see OESzmapResults.GetProbabilities).
Hence, the first (orderArray[0])
is the index of the orientation with the greatest probability
(probArray[orderArray[0]]).
The number of values in the output orderArray or the iterator is
OESzmapResults.NumOrientations.
order = rslt.GetProbabilityOrder()
print("conf with greatest prob = %d" % order[0])
Returns false or an empty iterator if this OESzmapResults is uninitialized.
NumOrientations
unsigned int NumOrientations() const
Returns the number of orientations for the probe molecule used in the calculation.
Equals the number of values returned by calls to
OESzmapResults.GetComponent,
OESzmapResults.GetProbabilities, or
OESzmapResults.GetProbabilityOrder.
PlaceNewAtom
OEChem::OEAtomBase *PlaceNewAtom(OEChem::OEMolBase &mol,
unsigned int element=OEChem::OEElemNo::O) const
Adds a new atom to the input molecule with atomic coordinates
of the 3D point provided to OECalcSzmapResults
when the object was created.
amol = oechem.OEGraphMol()
atom = rslt.PlaceNewAtom(amol)
print("vdw = %s" % atom.GetData("vdw"))
The new atom has been annotated with ensemble values for this point as generic data. String versions of the data have been formatted to two decimal places for convenient display.
Generic Data Tag |
Type |
Value (energies in kcal/mol) |
|---|---|---|
szmap_neut_diff_free_energy |
double |
Probe - neutral probe free energy difference |
szmap_order |
double |
Fractional entropy loss from electrostatics |
szmap_vdw |
double |
Van der Waals energy |
free-energy |
string |
Formatted szmap_neut_diff_free_energy |
order-param |
string |
Formatted szmap_order |
vdw |
string |
Formatted szmap_vdw |
The atom type can be controlled through the optional element parameter,
which defaults to oxygen.

Atoms Placed at Calculation Points with Generic Data Annotation
Returns a pointer to the newly created atom to facilitate further customization.
PlaceProbeMol
bool PlaceProbeMol(OEChem::OEMolBase &outputMol, unsigned int orientation=0u,
bool annotate=true) const
Modifies the outputMol to be a copy of one probe orientation,
placed at the 3D point provided to
OECalcSzmapResults when the object was created.
The probe orientation can be controlled through
the optional orientation parameter (the default value of 0 refers to
the first probe conformation).
pmol = oechem.OEGraphMol()
rslt.PlaceProbeMol(pmol, order[0])
If the optional parameter annotate is true (the default),
the molecule will be annotated with OEComponent
data for that orientation. See OESzmapResults.PlaceProbeSet
for more information on this annotation.
Returns false if this OESzmapResults is uninitialized.
PlaceProbeSet
double PlaceProbeSet(OEChem::OEMCMolBase &probeSet, double probCutoff,
bool clear=true) const
double PlaceProbeSet(OEChem::OEMCMolBase &probeSet, unsigned int maxConfs=0u,
bool clear=true) const
Modifies the multi-conformer probeSet to contain one or more orientations of
the probe, each placed at the 3D point provided
to OECalcSzmapResults when the object was created.
The probe set is returned in probability order
(see OESzmapResults.GetProbabilityOrder).
There are three ways to select which probe orientations are placed in the probeSet:
If just the
probeSetparameter is provided, without other options, all probe orientations will be returned.mcmol = oechem.OEMol() rslt.PlaceProbeSet(mcmol)
If the real number parameter
probCutoffis used, probe orientations will be added until the total cumulative probability is at least that amount. Cumulative probabilities are > 0.0 and <= 1.0.probCutoff = 0.5 rslt.PlaceProbeSet(mcmol, probCutoff) print("nconf to yield 50pct = %d" % mcmol.NumConfs())
Finally, if the integer parameter
maxConfsis used, no more than number of probe orientations will be returned. A value of0is a special signal to return all orientations.clear = False cumulativeProb = rslt.PlaceProbeSet(mcmol, 10, clear) print("best 10 cumulative prob = %.3f" % cumulativeProb)
If the optional parameter clear is set to false, any previous orientations
in the probeSet will not be cleared, allowing conformers for multiple 3D points
as well as multiple orientations to be stored in the probeSet. By default, previous
orientations are cleared away before the new orientations are added.
Each orientation has been annotated with OEComponent
data for that orientation.
In addition, the total interaction + psolv + wsolv + vdw energy of each
is recorded as the energy of the conformation
(accessible using the GetEnergy() method of the conformer).
String versions of the data
have been formatted to two decimal places for convenient display.
String data is also
stored as SD data, so they are included in VIDA’s spreadsheet
and can be saved to .sd files.
Generic/SD Data Tag |
Type |
SD |
Value (energies in kcal/mol) |
|---|---|---|---|
szmap_interaction |
double |
no |
Poisson-Boltzmann probe|context interaction |
szmap_psolv |
double |
no |
(Protein) context desolvation penalty |
szmap_wsolv |
double |
no |
(Water) probe desolvation penalty |
szmap_vdw |
double |
no |
Van der Waals energy |
szmap_probability |
double |
no |
Boltzmann probability |
total-energy |
string |
yes |
szmap_interaction + psolv + wsolv + vdw |
interaction |
string |
yes |
Formatted szmap_interaction |
psolv |
string |
yes |
Formatted szmap_psolv |
wsolv |
string |
yes |
Formatted szmap_wsolv |
vdw |
string |
yes |
Formatted szmap_vdw |
prob |
string |
yes |
Formatted szmap_probability |
Returns the cumulative probability of all the orientations returned, or 0.0 if this OESzmapResults is uninitialized.
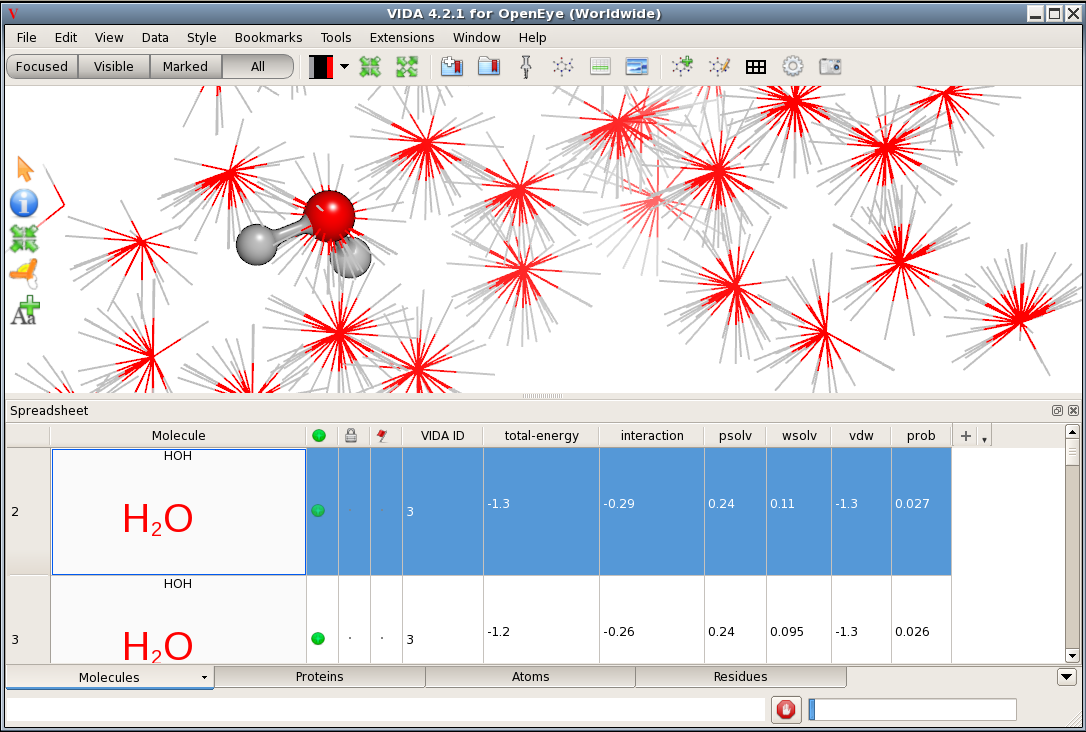
SD Annotation in the VIDA Spreadsheet