CheckCff
Overview
This is a simple utility that will simply apply a color force field to one or more input molecules and output a report of color atoms added, the TYPE of the added atom and the corresponding input molecule atoms that matched the SMARTS defining that TYPE. Additionally, checkcff will output an OEB file containing molecules with the color atoms added. This file can be loaded into VIDA and the atoms labeled with Name to visually inspect which color atoms are being added.
Example Commands
By default, checkcff will use the ImplicitMillsDean color force
field. This can be over-ridden by using the -chemff option and
providing either a name of one of the built-in color force fields
(ImplicitMillsDean, ExplicitMillsDean, ImplicitMillsDeanNoRings, ExplicitMillsDeanNoRings) or the name of a user-defined
color force field file.
So to see which atoms are considered color atoms using the ImplicitMillsDean force field:
prompt> checkcff -in mymolecules.sdf
To use the ExplicitMillsDean force field to see which atoms are considered to be color atoms:
prompt> checkcff -in molecules.sdf -chemff ExplicitMillsDean
The run always generates a report CSV file and an OEB file that can be viewed in VIDA:
prompt> checkcff -in 4cox-neutral.sdf
And the view in VIDA might look a bit like:
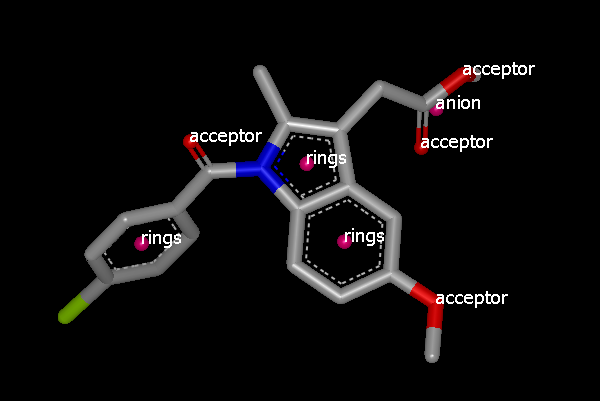
VIDA view of 4COX with color atoms attached and labeled.
Command Line Help
A description of the command line interface can be obtained by executing CheckCff with the –help option.
> checkcff --help
will generate the following output:
Help functions:
checkcff --help simple : Get a list of simple parameters
checkcff --help all : Get a complete list of parameters
checkcff --help defaults : List the defaults for all parameters
checkcff --help <parameter> : Get detailed help on a parameter
checkcff --help html : Create an html help file for this program
checkcff --help versions : List the toolkits and versions used in the application
Required Parameters
checkcff only has 1 required commandline parameter.
- -in <filename>
Input molecule file to be colored. Can be any one of the molecule file formats described in Section The Query File.
Optional Parameters
- -out <oebfile>
Output OEB file name for 3D structures with color atoms named by the TYPE from the color force field.
[default = checkcff_cff.oeb]
- -report <csvfile>
Override prefix used to name report file.
[default = checkcff_rpt.csv]
File Options
- -log
The argument for this flag specifies the name of the log file. The level of detail for logfile information can be altered using the
-verboseflag. Generation of an output log may be disabled by providingnulas a argument in Windows and/dev/nullas an argument on Linux and macOS. [default =prefix_log.txt]
- -prefix
The argument for this flag defines the prefix to be used for various information and data files generated. Most important among these is the ‘prefix_parm.txt’ file which includes a copy of all the parameters used in the run. The prefix is also used to generate a default log file name if not explicitly specified with the -log flag. [default = <app_name>].
- -verbose
This is a boolean flag that controls the level of detail written to the log file. By default only minimal information is written to the log file. Verbose logging will cause more information to be written to the log file in order to follow behavior during program execution. [default = false]
- -molNames
This parameter takes a text file containing a list of molecule names (one name per line in the file). If this parameter is set then only molecules in the input file(s) (see parameter
-in) with names that match those in the text files will be read in.The general purpose of this flag is to provide an easy mechanism for reading a few specific molecule(s) that are contained in a large file, without having to extract those molecules by hand from the input molecules file. [default = NONE].
- -progress
Show progress on screen. Options are ‘none’, ‘dots’, ‘log’ and ‘percent’. The ‘dots’ options will displays dots on screen to show molecules completed. The ‘log’ option will duplicate the log file on screen. The ‘percent’ option will track progress through the input file. [default = none]
Overlap Prep Option Parameters
There are 6 overlap prep option parameters.
- -chemff <cff_file>
Color-force-field name. Either the name of one of the built-in color force fields (ImplicitMillsDean or ExplicitMillsDean or ImplicitMillsDeanNoRings or ExplicitMillsDeanNoRings) or the name of a user-defined color force field file. The format of this file is given in section Color Force Field.
[default = ImplicitMillsDean]
- -assignColor
Determines the state of the assign color flag. If true, color atoms would be assigned during the Prep.
[default = true]
- -useHydrogens
Determines the state of the use hydrogens flag. If true, explicit hydrogens atoms would be created during the Prep. If false, all hydrogen atoms would be removed.
[default = false]
- -assignBondiRadii
Determines the state of the assign Bondi radii flag. If true, Bondi radii would be assigned to all the atom during the Prep.
[default = false]
- -removeDups
Determines if close color atoms with the distance less than -minDistance should be removed.
[default = false]
- -minDistance
Threshold for duplicate removal. A value in the interval (0,1) that determines the minimum allowed distance between two color atoms.
[default = 0.001]