Release Highlights 2022.2.2
The OEApplications 2022.2.2 is a bug-fix of the OEApplications 2022.2.1 release, and depends on OEToolkits 2022.2.2.
section_szybki_releasenotes_2512
Release Highlights 2022.2.1
MCS based fix during OMEGA Conformer Generation
The ability to constrain a fragment of the molecules during conformer generation with torsion
driving in OMEGA and the OMEGA toolkit
has been extended to include fixing based on maximum common
subgraph (MCS) match. In this mode of fixing, the MCS between the template fixmol and the generated
molecule is determined, and subsequently that common subgraph portion of the generated molecule is
fixed to the proved conformer of the fixmol.
The MCS based fixing during conformer generation can be turned on by setting
SetFixMCS to true for use in the
OMEGA toolkit. Additional
API points have also been added to allow control during MCS search. A new flag -fixMCS is
available in the OMEGA application to facilitate this.
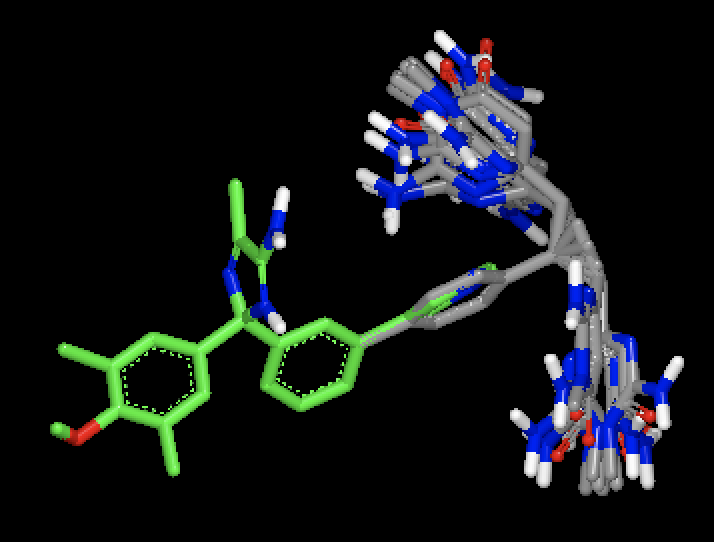
|
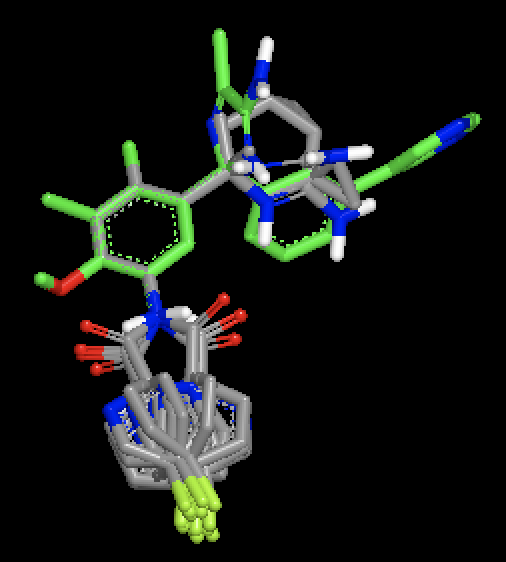
|
OMEGA generated conformers for two different molecules, with MCS based fix against a single bound ligand pose. Figure shows that with MCS based fix, two different fragments are fixed during conformer generation of the two different molecules.
New ShapeFit Algorithm for Pose Prediction
The algorithm for pose prediction with the SHAPEFIT
method has been modified. The new algorithm simultaneously optimizes the shape and
chemical similarity between the fit molecule and bound ligand, and intra-molecular
force-field energies of the fit molecule. The new simultaneous optimization algorithm
replaces the previous adiabatic optimization algorithm and is more robust. The new
algorithm also offers flexibility to use different forcefields and uses the
SAGE force field by default. Another advantage of the new
algorithm is that it is capable of producing multiple distinct poses when desired.
The new algorithm is automatically reflected in both the OEPosit toolkit
and the POSIT application. ShapeFit is also now available in the
OEDocking TK as an independent API OEShapeFit.
Cross docking experiments based on the 22 diverse kinase types presented in
[Tuccinardi-2010] shows that ShapeFit accuracy is unaffected between the
previous and the new algorithm (when accuracy is measured as RMSD <=2.0).
A balanced cross-docking set of 20,000 points, where points are equally distributed
over the various similarity regions and kinase types, was used. However, we see
clear indication that generating multiple poses helps increase the pose prediction accuracy.
A separate self-docking study based on the highly trustworthy iridium dataset ([Warren-2012])
with 280 ligand-protein structures shows that new algorithm predicts poses that are
closer to the x-ray crystallographic poses.
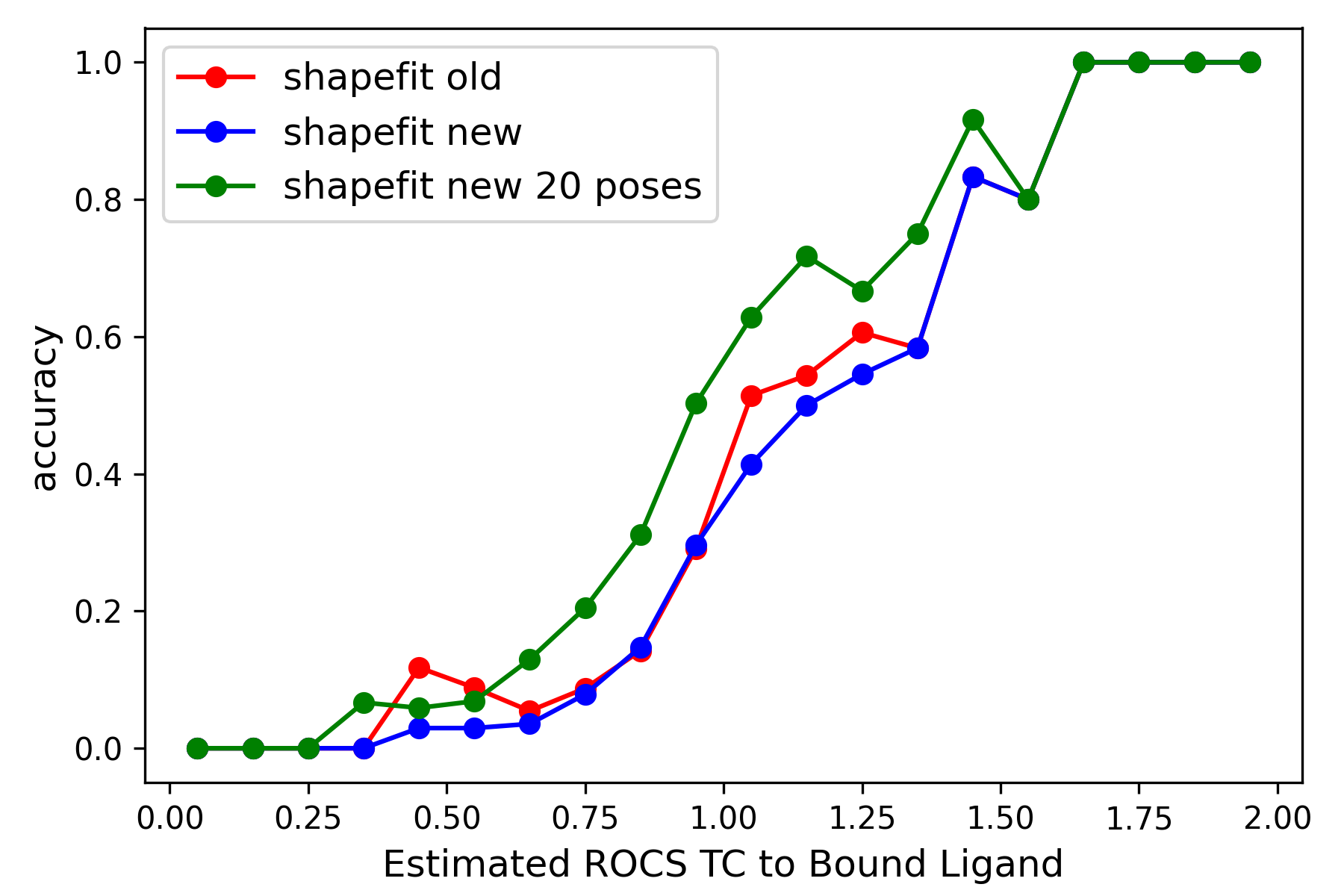
|
|
Left: Comparison of ShapeFit accuracy in a cross-docking experiment between the existing and the new algorithm. The x-axis shows the tanimotocombo similarity between the ligand and the bound ligand present in protein complex. The Y-axis shows the fraction of accurately predicted poses based on being within 2 angstroms of the experimental pose. Right: Comparison of ShapeFit accuracy in a self-docking experiment, between the existing and the new algorithm. Axes show the RMSD of generated poses with respect to the experimental pose.
Protein-ligand Optimization with ff14SB forcefield and PB solvent model
Protein-ligand Optimization with ff14SB-OpenFF force field and Poisson-Boltzmann (PB) solvent model
is now available both in SZYBKI and the SZYBKI toolkit.
In the SZYBKI TK, the functionality is available through the OEFixedProteinLigandOptimizer
and the OEFlexProteinLigandOptimizer APIs, and can be enabled by setting
PB as the value in SetSolventModel.
In SZYBKI both the OptLigandInDU and OptimizeDU applications now accept pb as value
for -solventModel.
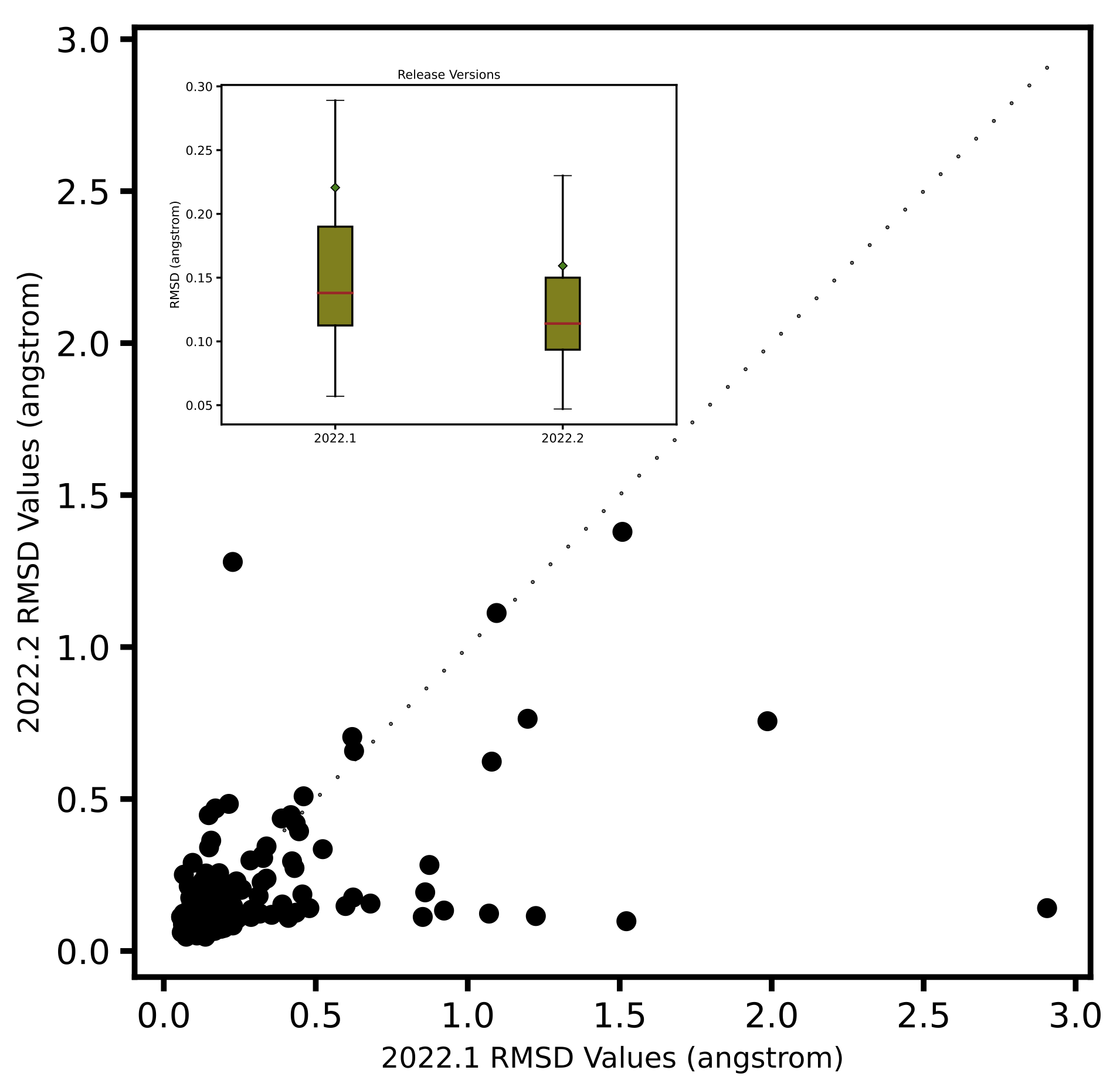
Optimization of 244 highly trustworthy x-ray structures of protein-ligand complexes from the
iridium dataset ([Warren-2012]) with and without PB solvation
and with ff14SB-Sage forcefield shows better accuracy for the optimized ligand poses when PB solvation
is turned on. Accuracy is measured as the RMSD between the crystallographic ligand pose and the optimized
pose. On an average PB solvation optimized structures have an RMSD of 0.31 angstroms, compared to a value of
0.55 angstroms when explicit solvation is not turned on. A paired T-test suggests that the average improvement
of 0.24 angstroms is statistically significant with a p value of 0.00001. Turing on the solvation effects also
removes any significant deviations (RMSD > 2 angstroms) of pose, as was seen for a few cases when optimization was
performed in vacuum.
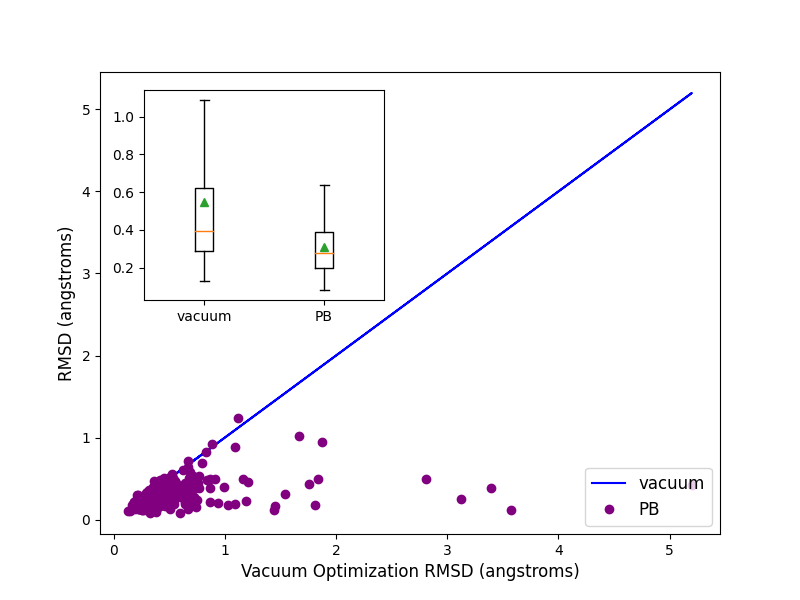
Comparison of RMSD for Flexible protein-ligand optimization of x-ray structures with and without PB solvation and with ff14SB-Sage forcefield.
New BROOD Fragment Databases
Two new BROOD fragment databases, brood-database-ChEMBL31 and brood-database-ChEMBL31_lite,
built from the latest version of ChEMBL, have been generated and are made available. The
brood-database-ChEMBL31 contains all possible fragments up to 3 attachment points, whereas
the brood-database-ChEMBL31_lite is curated to prioritize fragments with medicinal relevance.
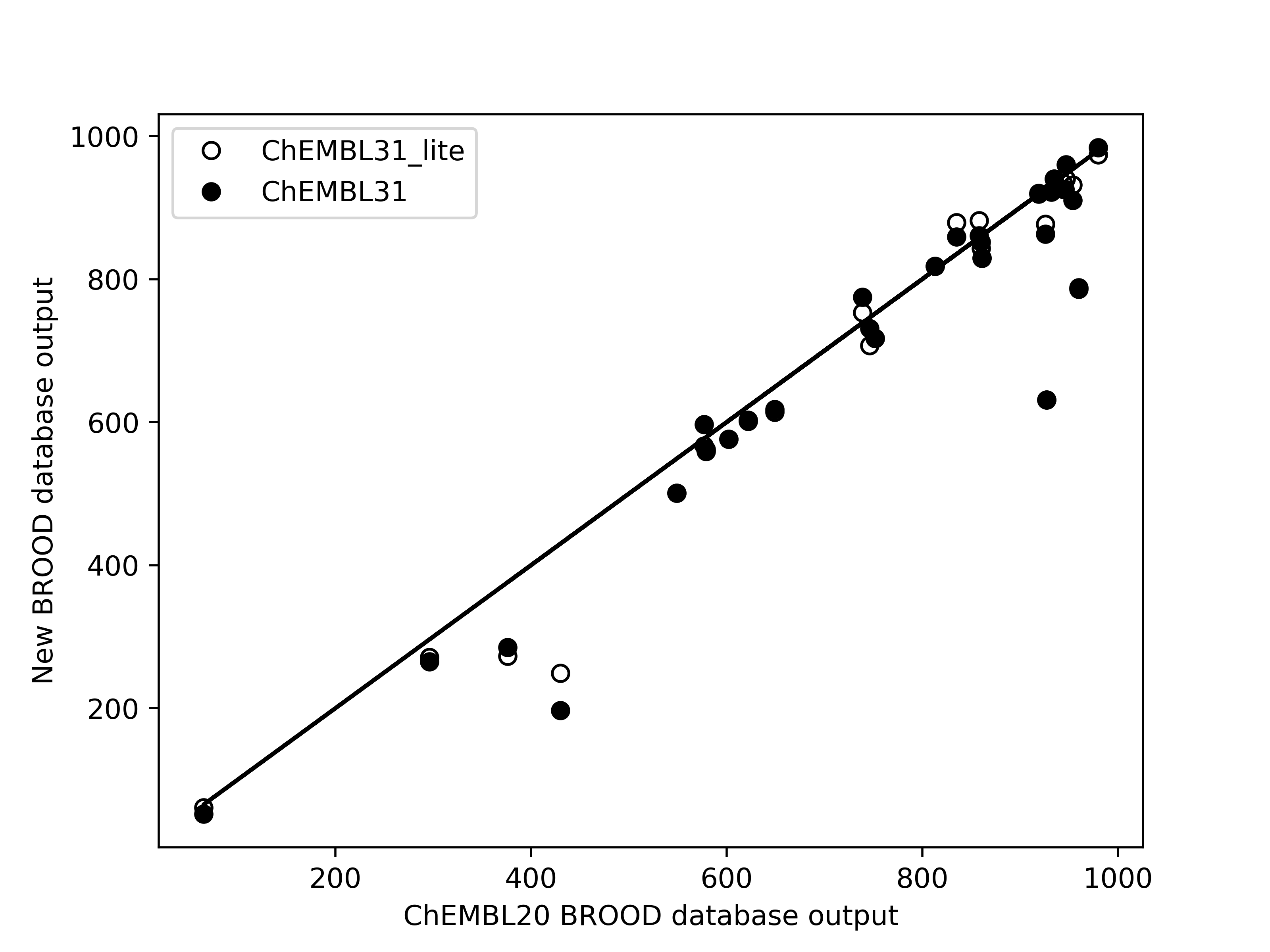
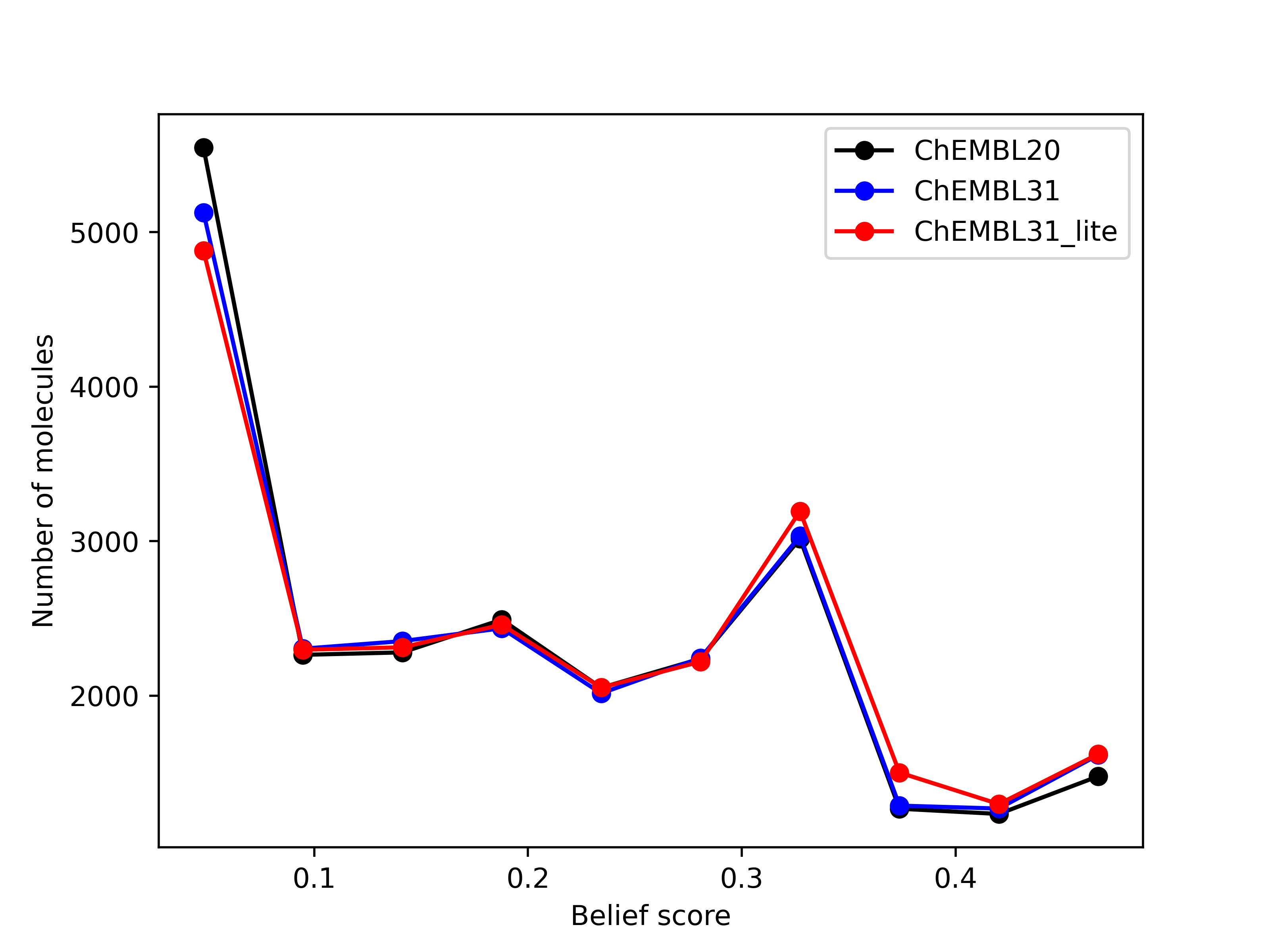
Limited validation, with 29 different queries across 14 molecules, shows that the number of hits of
druglike molecules obtained from the new databases are within 1% of those generated from the existing
database brood-database-chembl-3.0.0. Comparison of belief score, a measure of potential activity
of the generated hits, also shows that the hits generated from the new databases are at least as
good as those generated from the existing database.
|
|
Left: Comparison of the number of hits of druglike molecules using the brood-database-chembl-3.0.0 (ChEMBL20), brood-database-ChEMBL31 (ChEMBL31) and brood-database-ChEMBL31_lite (ChEMBL31_lite) fragment databases. Right: Comparison of the belief score distribution of all the hits generated using the ChEMBL20, ChEMBL31 and ChEMBL31_lite fragment databases.
Supported Platforms
OS
Versions
Linux
RHEL7/8, Ubuntu20/22
Windows
Win10, Win11
macOS
11, 12
General Notices
Support for Ubuntu22 has been added. Support for Ubuntu18 has been dropped.
This is the last release to support macOS 11. Support for macOS 13 will be added in the next release.
This is the last release to support RHEL7. Support for RHEL9 will be added in the next release.