Release Highlights 2021.2
SiteHopper: New Toolkit for Protein Binding Site Comparison
The 2021.2 release introduces SiteHopper TK, a new toolkit for searching a database containing design units for proteins with similar binding sites to a query design unit. The new SiteHopper TK provides toolkit level access to the SiteHopper application functionality released in 2021.1.
Besides providing toolkit level access, the 2021.2 release also brings several other improvements to SiteHopper. Efficiency of database building in SiteHopper is enhanced with the use of a multi-threaded approach. Furthermore, search is now possible on the CPU, enabling cross platform support of the toolkit and application. On Linux machines with an NVIDIA graphics card, the GPU is used as a pre-screening method, speeding up the search dramatically. A feature to exclude results based on a similarity threshold has been added that allows for more diversity in the protein targets with similar binding sites. The database storage format has also been revised.
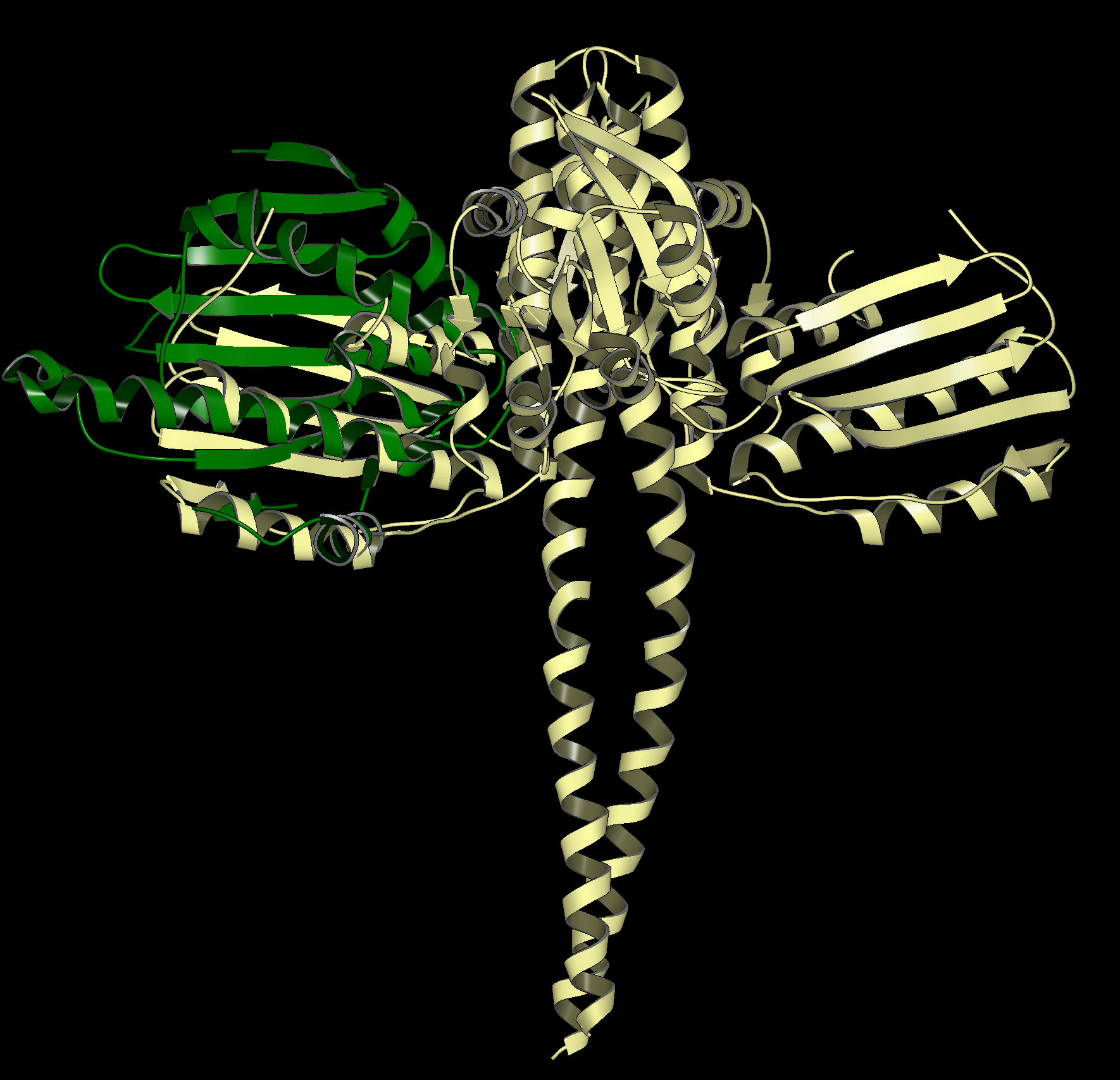
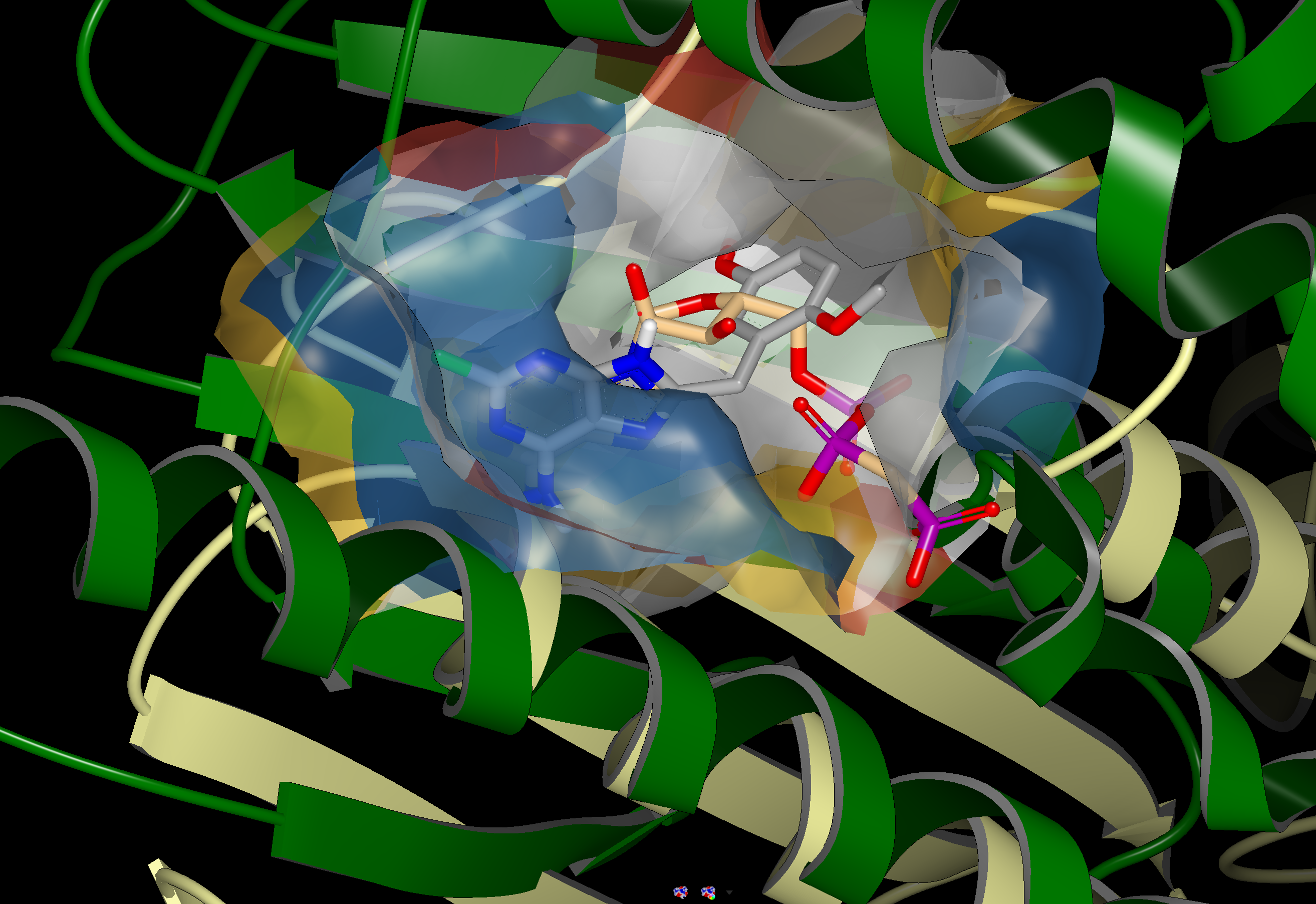
An example of a SiteHopper produced hit is shown in the figure below. 1UYG is a structure of human heat shock protein 90-alpha, bound to 8-(2,5-dimethoxy-benzyl)-2-fluoro-9H-purin-6-ylamine. 5IUN is a structure of the DesK-DesR complex, bound to AMP-PCP. The image on the left shows the overlay of 1UYG (green) and 5UIN (light yellow), zoomed out to show the major structural differences between the two proteins. The image on the right zooms in on the binding sites, with a surface showing the type of residues present in each binding site. Blue represents acidic, red represents basic, yellow represents polar, and white represents non polar. Despite a sequence similarity of only 46%, 1UYG and 5IUN may be targetable by similar ligands, as they have very similar steric and electrostatic properties in their binding sites.
|
|
1UYG human hsp 90 (green) overlaid with 5UIN N-formyltransferase (yellow), full struture view (left), binding site view (right).
SZYBKI: OpenFF-Sage force field support
The latest force field, Sage, from the Open Force Field Initiative has been added to OEFF TK, Szybki TK, and SZYBKI. The major features of the Sage force field are outlined here.
New force field parameter classes OESageParams and OEFF14SBSageParams have been added to the toolkit to support the Sage parameters. Three new force field classes, OESage, OEFF14SBSage, and OESage, have also been introduced. The Sage force field is now the default for free energy analysis of conformation ensembles with OEFreeFormConf, and the corresponding application Freeform. The protein-ligand forcefield, FF14SBSage, is now the default for protein-ligand optimization in OEFixedProteinLigandOptimizer and OEFlexProteinLigandOptimizer, OptimizeDU, and OptLigandInDU.
The Sage force field can be used in SZYBKI for ligand calculations, with
the -ff sage_openff2.0.0 command line option, and for calculations involving proteins using
-ff ff14sb_sage.
OEChem: MMCIF and CIF writers
Functionality to write Crystallographic Information Files (CIF and MMCIF) has been added to the OEChem TK in the 2021.2 release. Similar to writing other molecule file formats in OEChem TK, CIF and MMCIF files can be written using the high level WriteMolecule function. Low level functionality for advanced users is also provided through a new OEWriteCIFFile function.
If the molecule does not contain residue information, it is written as a small molecule CIF file if the data is available to do so (i.e. space group information). If residue information is available, the writer checks the number of residues and the molecule is either written as a chemical component dictionary entry (in the event there is a single residue) or as a macromolecular CIF file (MMCIF).
The small molecule CIF reader has also been improved for performance and robustness, and round-tripping these files has been tested and validated using the checkCIF facility from the International Union of Crystallography (IUCR). The MMCIF parser based on Gemmi has also been updated, and items parsed from PDB metadata have been expanded for proper processing of the biomolecules with Spruce.
Supported Platforms
OS
Versions
Linux
RHEL7/8, Ubuntu18/20
Windows
Win10
macOS
10.14, 10.15, 11
General Notices
This is the last release to support macOS 10.14. Support for macOS 12 will be added in the next release.
Support for Windows 11 will be added in the next release.