Sequence-to-Structure Tutorial
The sequence-to-structure prediction floes use the Boltz model for macromolecular structure prediction and then Spruce to prepare them for downstream modeling applications. These floes are highly customizable, and this tutorial covers various sequence input methods and the multiple sequence alignment (MSA) search options.
Floes Used in This Tutorial
The floes used in this tutorial are:
Tutorial: Multiple Sequence Alignment
A multiple sequence alignment is commonly used in conjunction with Boltz structure prediction to increase accuracy. The Protein Sequence to AI Folded Structure Prediction Floe can be run with an MSA search step that runs prior to structure prediction. However, for this tutorial, the MSA Align and Search Floe will be used to generate an MSA search result which is then used as input to the structure prediction floe.
Locate the Floe in Orion
Navigate to the Floe page using the blue navigation bar.
On the Floe page, click on the Workfloes Tab, where you will find a list of the available floes and packages.
Under the Filters in the left-hand pane, click on the caret next to the Packages filter to expand the list of packages and click on the OpenEye AI Fold Floes package. This will ensure that the floes listed in the middle pane are from this package.
From this list, select the MSA Align and Search Floe, then click the blue “Launch Floe” button to launch the Job Form.
Provide Input Files and Parameters to Run the Floe
Sequences can be input using either string inputs or a FASTA file input. This tutorial covers the FASTA file input strategy. The FASTA file first needs to be uploaded to Orion and then can be selected as a file input in the Job Form.
Multiple sequences can be entered as input so long as their sequence titles are unique. The expected format of sequence titles in the FASTA file follow the RCSB format. However, as long as the text prior to the first ‘|’ delimiter is unique, the search will not detect duplicate sequences. For example, a FASTA title of ‘>2MG4_1|Drosophila melanogaster’ will be automatically assigned the 2MG4_1 sequence title.

Figure 1. MSA input on the Job Form.
The Input MSA Collection is a collection of FASTA sequences. Several commonly used MSA databases have already been prepared and can be found in the AIFold directory in the OpenEye Data folder of Organization Data. Custom collections can also be created using the MSA Collection Setup from FASTA Floe and used as input.
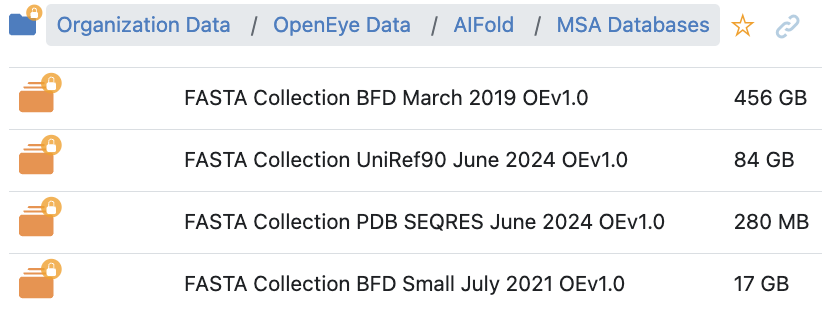
Figure 2. The list of MSA FASTA collections available in Orion.
MSA Results
MSA results are saved to an output .a3m file in your output directory and will be used as input for the Protein Sequence to AI Folded Structure Prediction Floe. Each unique sequence in an input will have its own MSA search result keyed on the input sequence title. This key is what is used in subsequent floes to match an MSA to a sequence for structure prediction.
Tutorial: Sequence-to-Structure
The structure prediction floes use the Boltz model to run structure prediction and expose many features from this model in the floe’s Job Form. The Protein Sequence to AI Folded Structure Prediction Floe and the related Protein Sequence to AI Folded Structure Ligand Affinities Floe allow for only one protein system to be predicted per job. Multiple sequences in the inputs indicate multimeric protein folding.
Provide Input Files and Parameters to Run the Floe
On the Floe page, locate the Protein Sequence to AI Folded Structure Prediction Floe, then click the “Launch Floe” button to launch the Job Form.
Sequences should be entered as described in the Multiple Sequence Alignment section, and special consideration should be given to sequence titles. In various sections of the Job Form (multimers, constraints, MSA, etc.), you will need to know this sequence title to properly apply a feature. This title is defined to be the text between the FASTA title (‘>’) and the first pipe delineator (‘|’). For example, a FASTA title ‘>2MG4_1|Drosophila melanogaster’ will be automatically assigned the 2MG4_1 sequence title.
Chain identifiers are used to explicitly identify a sequence’s chain or to indicate whether multiple instances of the same chain (e.g., monomeric dimer) are to be co-folded. You can use the sequence title to identify the chain and unique strings to identify the chain ID. It is acceptable to leave this field blank and only one instance of the input chain will be used during structure prediction. For this example, you want to fold a monomeric dimer, so populate this field with the value: 2MG4_1:A,B.
Ligand co-folders are identified with a SMILES string or CCD molecule ID, followed by a unique chain ID. This example does not contain a co-folded ligand, so leave this field blank.
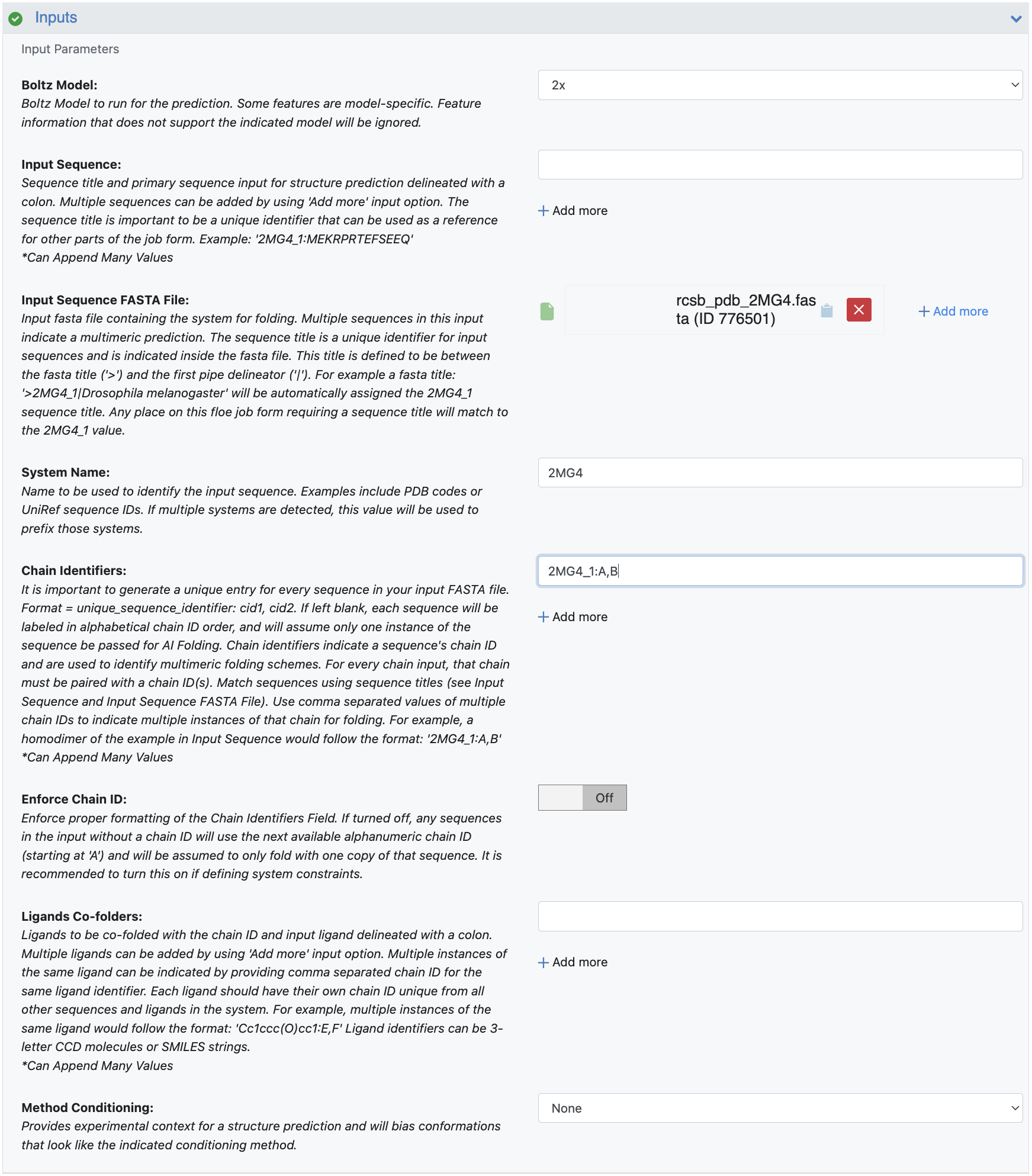
Figure 3. Sequence-to-structure input parameters.
The MSA search options are where you should provide the results from the Multiple Sequence Alignment section to this structure prediction. The MSA Align and Search Floe will generate a unique .a3m file for each unique input sequence, all of which need to be provided to the structure prediction floe. This system only contains one unique sequence and will be added to the Precomputed MSA File parameter field.
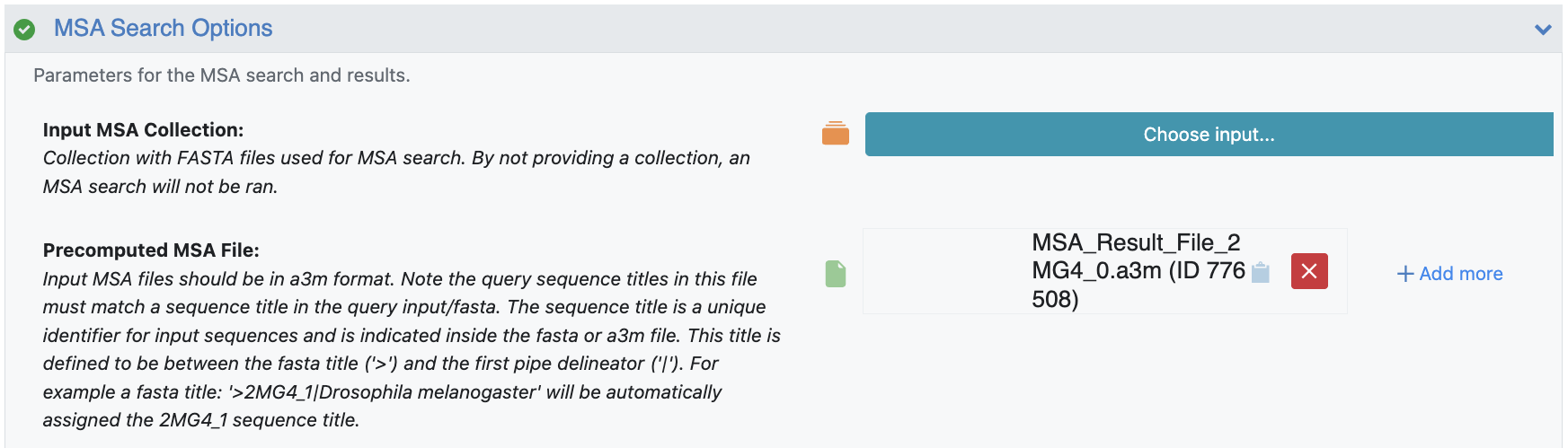
Figure 4. MSA Search Option parameters.
After providing the input sequences and inputs, click on the green “Start Job” button.
Visualize AI Folded Structures
Once the Protein Sequence to AI Folded Structure Prediction job has finished, two datasets may have been generated. The dataset generated by the Boltz Mol Results parameter will contain the structure predictions straight from Boltz, and the dataset generated by the Boltz Results parameter will contain the Boltz structure predictions that have been standardized using Spruce preparation and formed into a design unit.
To visualize the prepared dataset, make the Spruce-prepared active as follows:
On the Data page, clear current active datasets by clicking on the Active Datasets drop-down and then “Clear All.”
Activate the output datasets by clicking on the + icon (“Dataset Activation” button).
The 3D results can be visualized on the 3D & Analyze page, and the spreadsheet shows the various prediction and confidence metrics provided by Boltz. The spreadsheet will also contain property predictions if these were indicated in the Job Form (e.g., affinity).
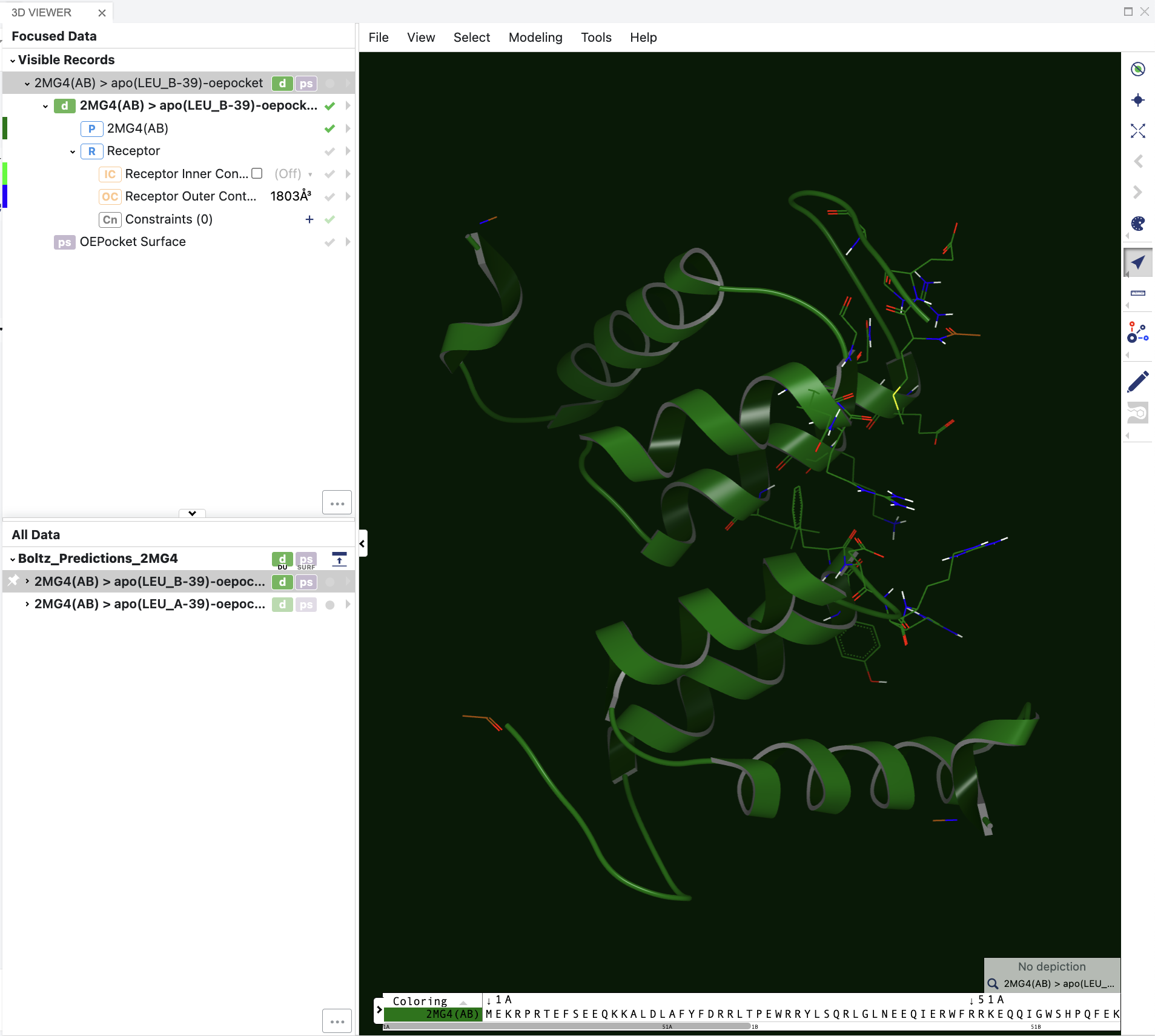
Figure 5. The 3D visualization of the sequence-to-structure results.
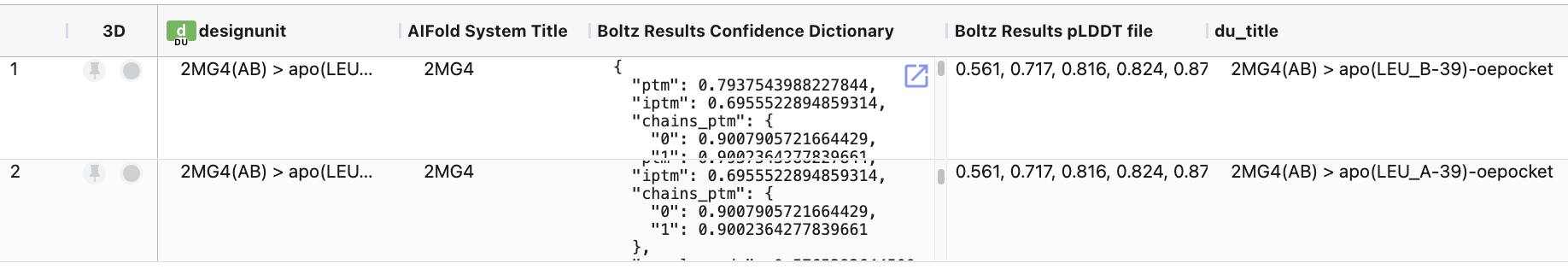
Figure 6. The spreadsheet showing the sequence-to-structure results.