Tutorial 6: NGS UMIs Extract and Annotation Floe
Background
A selection campaign using a semi-synthetic, codon-optimized, antibody library was carried out against the SARS-COV-2 antigen Spike(S) trimer and its subunit S1 and the S1 subunit RBD. Three independent selection campaigns were carried out using the same input library. DNA was isolated from a less stringent population, 2 rounds of in-vitro selection at 10nM, and a more stringent population, 3 rounds of in-vitro selection at 1nM. Each population was prepared by PCR amplification with the addition of in-line barcode consisting of 8mer (DNA) barcodes at the 5’ and 3’ ends to distinguish the 6 populations consisting of:
1) 2 x trimer (early and late rounds)2) 2 x S1 (early and late rounds)3) 2 x RBD (early and late rounds)
All input files can be located in Orion in following location:
Organization Data / OpenEye Data / Tutorial Data / AbXtract /
A barcode table titled ‘barcode_file_abxtract.xlsx’ indicates how these samples were prepared. Amplified DNA products were pooled together in a single tube and shipped to a PacBio / Illumina sequencing provider. The provider then shipped us back one PacBio file titled ‘pacbio_small_codon_optimized.fastq’ and two illumina files titled ‘F_illumina_codon_optimized_abxtract.fastq and R_illumina_codon_optimized_abxtract.fastq’, respectively. The PacBio file consists of a set of sequences each of length ~800bp composed of variable light (VL) and variable heavy (VH) sequences in VL-linker-VH orientation. The Illumina files consist of one forward and one reverse FASTQ file. Each sequence within the FASTQ is ~300bp in length, with an overlapping region between the forward and reverse reads to enable paired end assembly of the sequences into one VH sequence.
For the purposes of this tutorial, each of the FASTQ files from PacBio/Illumina was reduced by taking a random output of 87,966 sequences for PacBio and 84,988 sequences from Illumina. The larger files from which these were derived contain an underlying diversity of full-length nucleotide of ~50k total, with a 20-fold reduction in the H3 diversity.
All populations are derived from the same input library against three overlapping subunits of the Spike (S) protein. The goal of this tutorial is to cluster all three populations binding to the Spike (S) trimer, which encompasses S1. S1 can further be broken into smaller units, one of which is the receptor binding domain (RBD). Because each domain is subunit of the other, we anticipate overlap among RBD, S1 and trimer, particularly since the same input library was used during this selection. We anticipate we will also find antibodies that are unique to select subunits (e.g. S1 only), likely due to 1) non-overlapping regions (e.g., epitopes on trimer outside of S1), 2) exposed regions that are available in recombinant form but not in complex (e.g., S1 in trimer complex versus S1 in recombinant form), 3) and conformational changes from native or complex form versus recombinant form.
The goal of the tutorial is to identify overlapping regions of interest (ROI) particularly at the heavy chain complementarity determining region (HCDR3), which is most often implicated in binding specificity of the antibody to the target. In particular, we will use the HCDR3 to identify the output populations that overlap across the RBD, S1 and trimer populations as we hypothesize the population at the intersection are likely to exhibit optimal binding profiles in the native conformation of the RBD in vivo, which will result in more optimal neutralization (RBD) potential relative to those HCDR3s that only recognize recombinant versions of the RBD or S1. Further, we want to ensure that antibodies display more favorable NGS metrics (high relative abundance, elevated fold enrichment, increased copy number of the full-length antibody, and reduced number of liabilities that may present problems during scaling up processes).
STEP 1 - Login to Orion, Set-Up Directory, Upload Files
If the sequencing provider provides FASTQ files that are compressed (e.g., ends with “.gz” or “.zip”), decompress with a standard decompressor so the file ends with .fastq. If any suffix is added to the end, make sure to modify these suffixes so they end in .fastq for all sequence files to be processed.
Log into the Orion interface with your email and password. Find the 3 required files under Organization Data as follows:
A) Organization Data / OpenEye Data / Tutorial Data / AbXtract / patient_tutorial.fastqB) Organization Data / OpenEye Data / Tutorial Data / AbXtract / patient_barcode.xlsCreate a general tutorial directory and tutorial 6 subdirectory under PROJECT DIRECTORY / TUTORIALS / TUTORIAL_6 (This is your BASE DIRECTORY and should be used for all outputs for this Tutorial 6 below).
STEP 2 - Select the ‘NGS Pipeline with Automated Lead Selection’ Floe
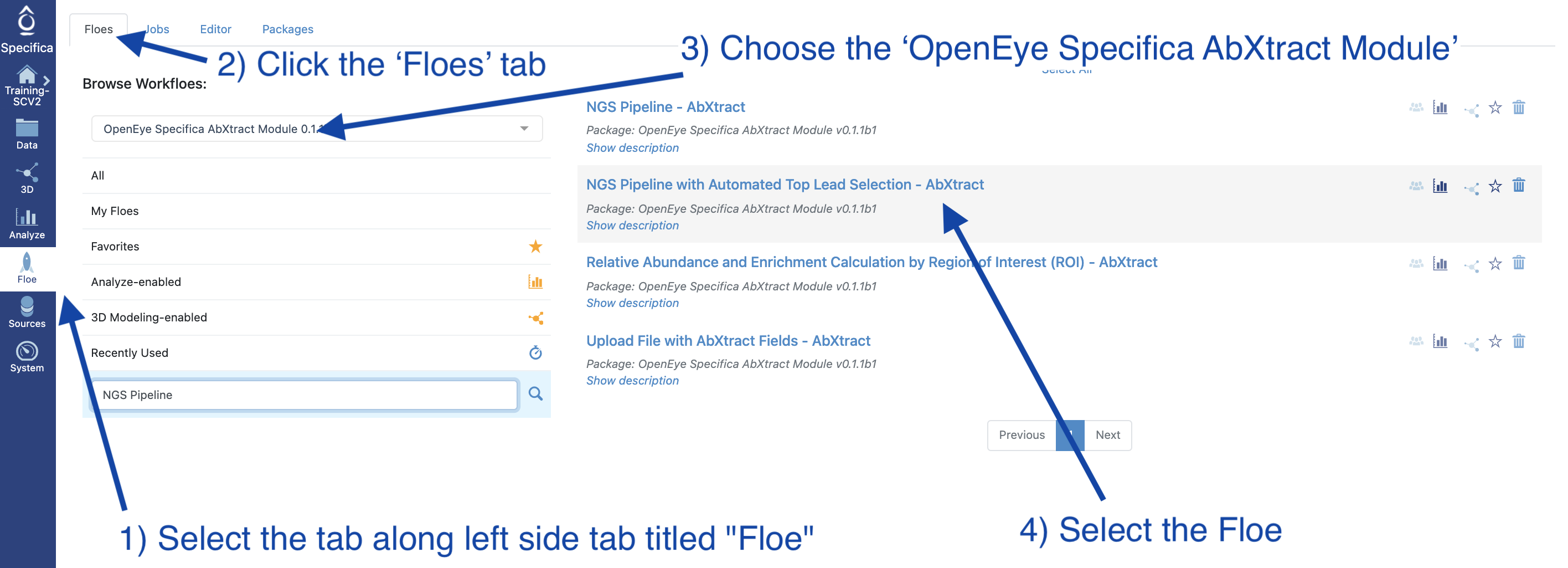
Select the tab along the left side tab titled ‘Floe’.
Click the ‘Floes’ tab.
Choose the ‘OpenEye Specifica AbXtract Module’.
Select the Floe ‘NGS Pipeline with Automated Top Lead Selection’.
STEP 3 - Prepare PacBio Run and Start Job
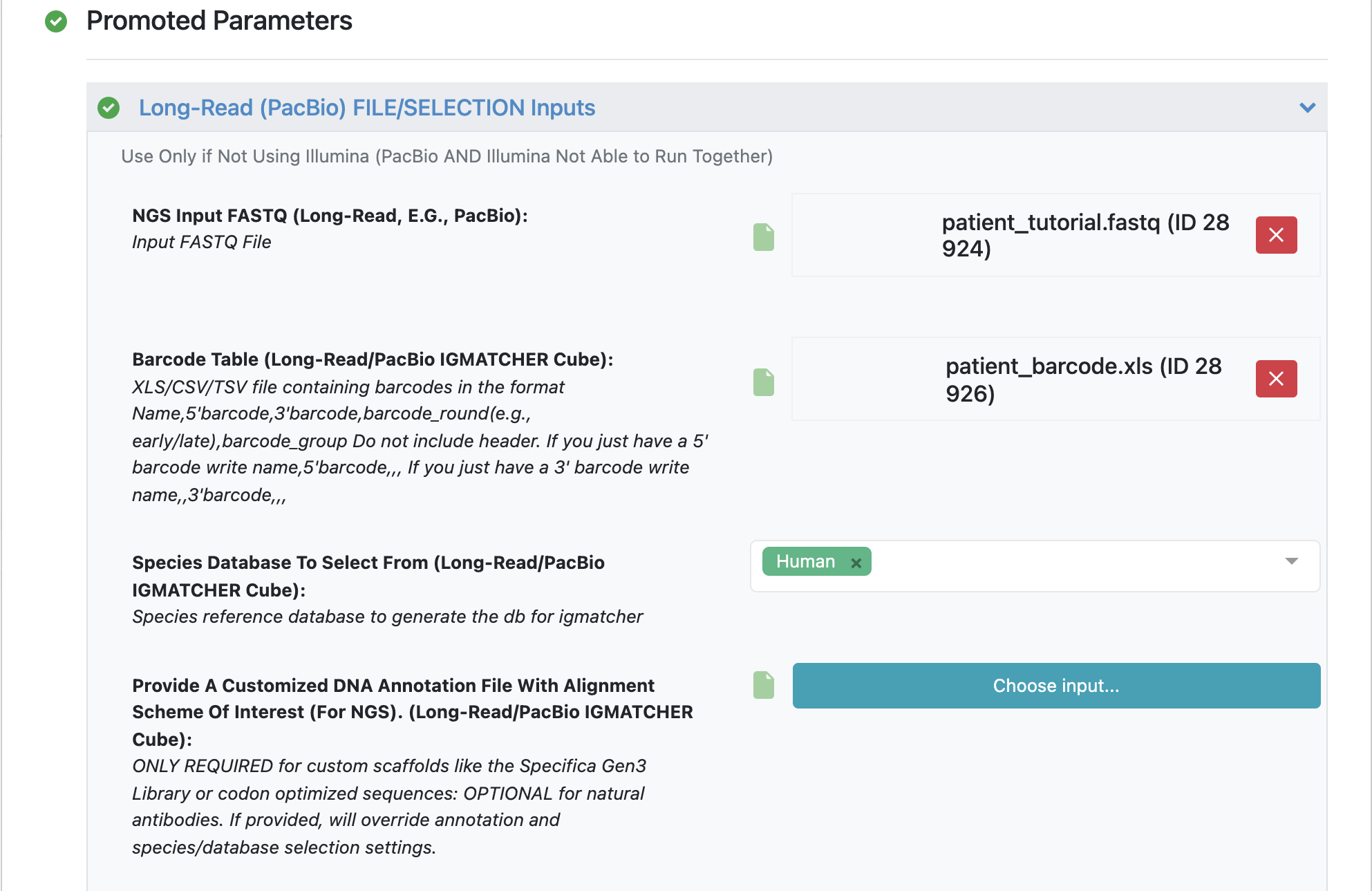
Make sure to specify (and remember) the directory output for all datasets and files.
Load FASTQ file - ‘patient_tutorial.fastq’.
Load barcode XLSX file - ‘patient_barcode.xls’.
STEP 4 - Select Different Clonotyping Method
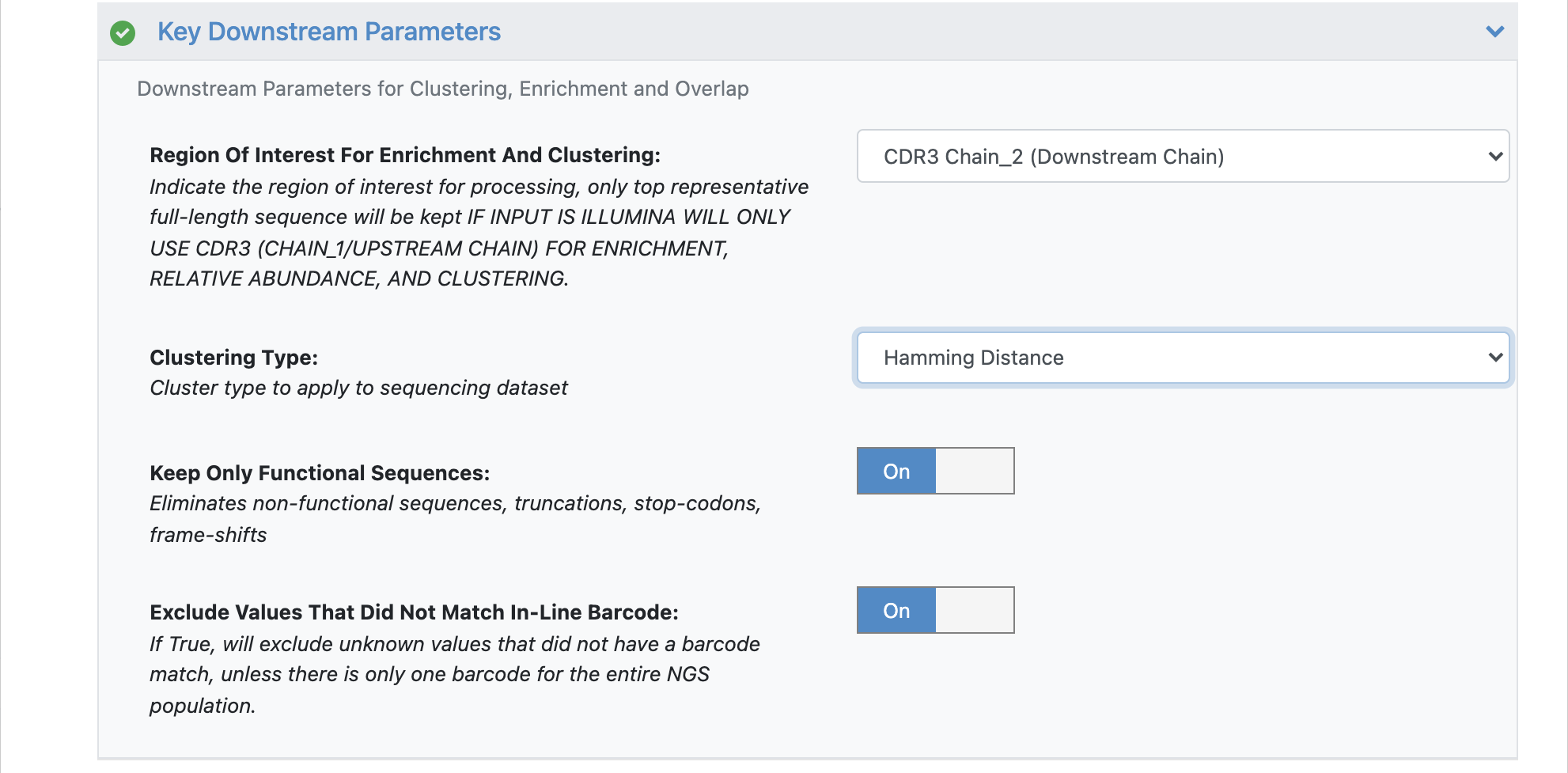
Under the promoted ‘Key Downstream Parameters’, click to expand.
Change the ‘Clustering Type’ to ‘Hamming Distance’.
In order to adjust the maximum Hamming distance (default = 2) we need to open the ‘Hidden Parameters’.
Turn the option ‘Show cube parameters’ to ‘Yes’.
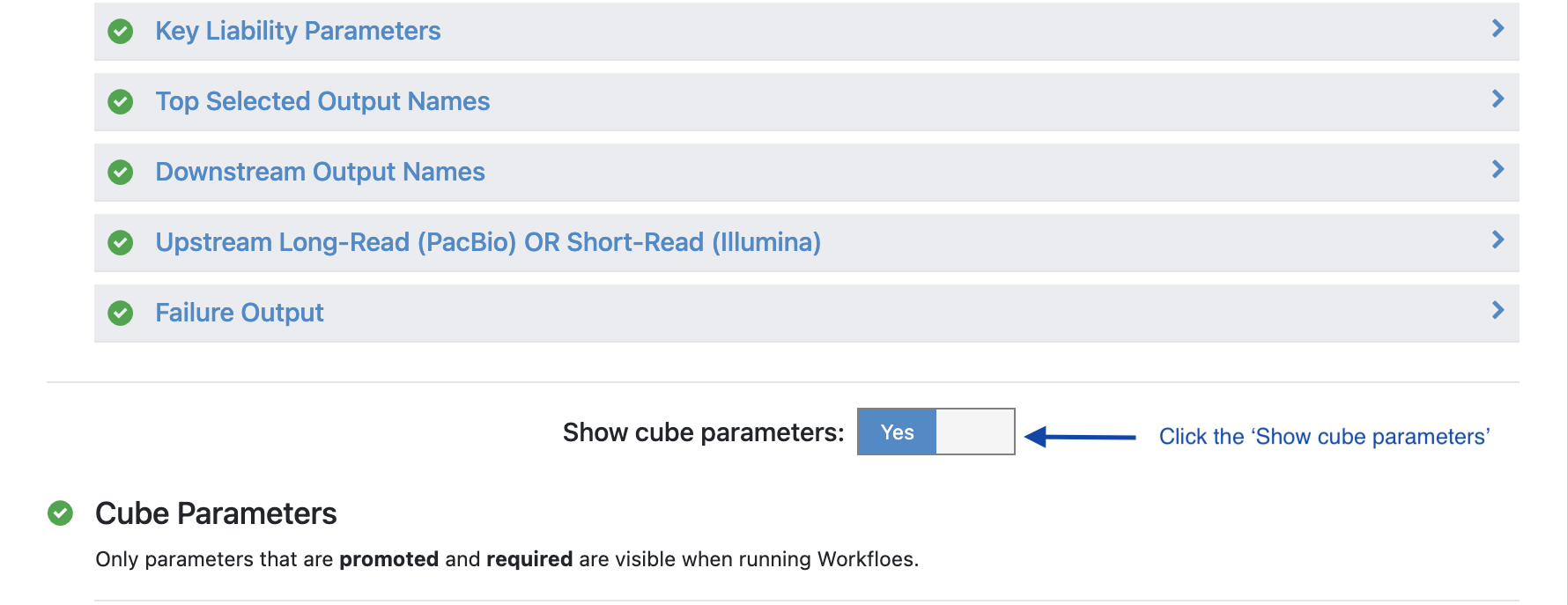
Under ‘Cube Parameters’ find the box titled ‘Downstream KEY PARAMETER Cube’. Click to expand.
Find the ‘Max Distance for Levenshtein or Hamming, If Selected’. Change this value from 2 to 4.
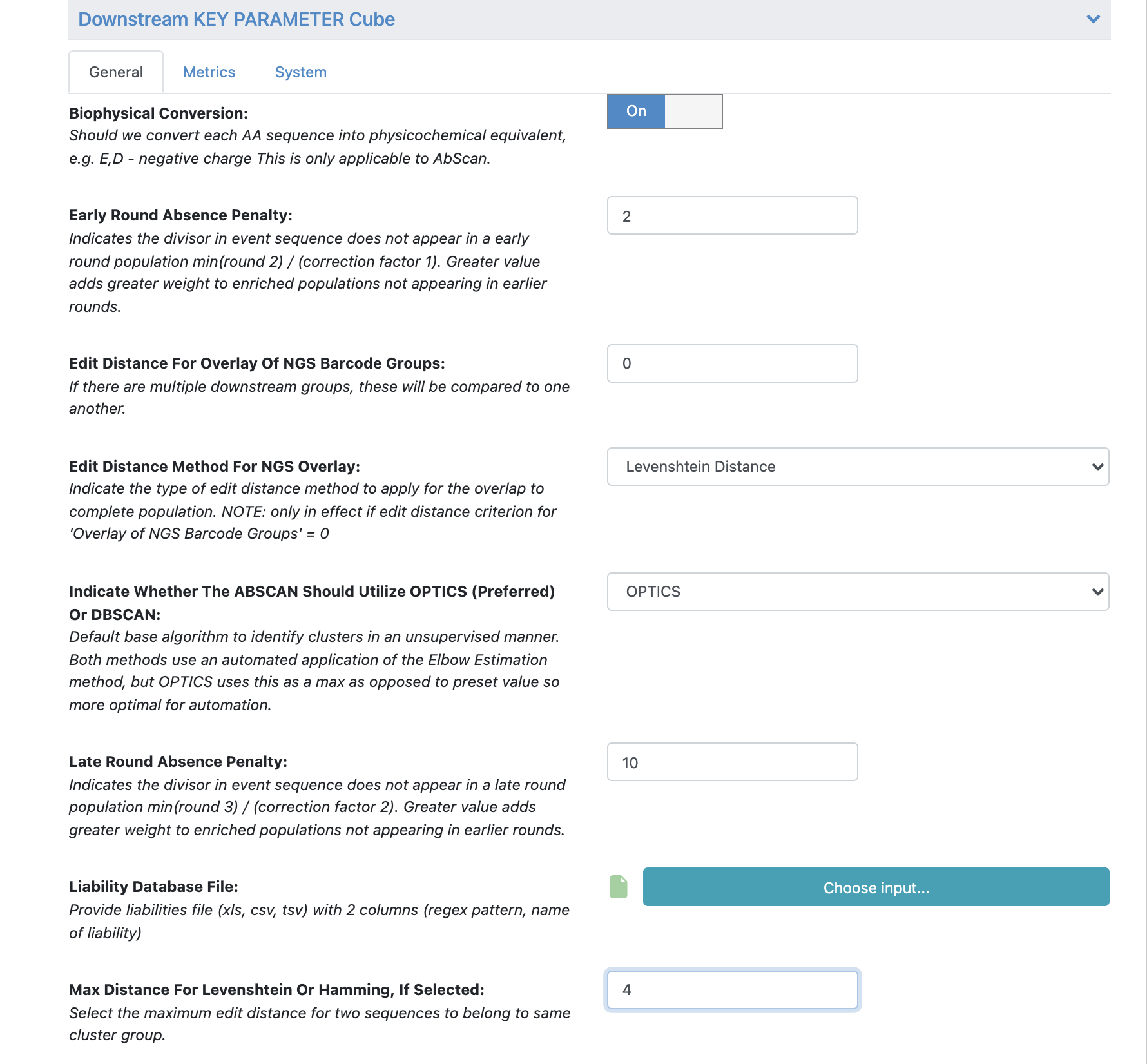
All the remaining parameters can be kept as the default values. Scroll through remaining “Promoted” parameters to understand these in greater detail.
See tutorial 2 automated selection options for more details.
Click ‘Start Job’.
STEP 5 - Identify both the Picked and the Downstream Population
Find the output directory specified above.
Identify the datasets labeled ‘picked.consolidated’ and ‘down.consolidated’ make both of these datasets active.
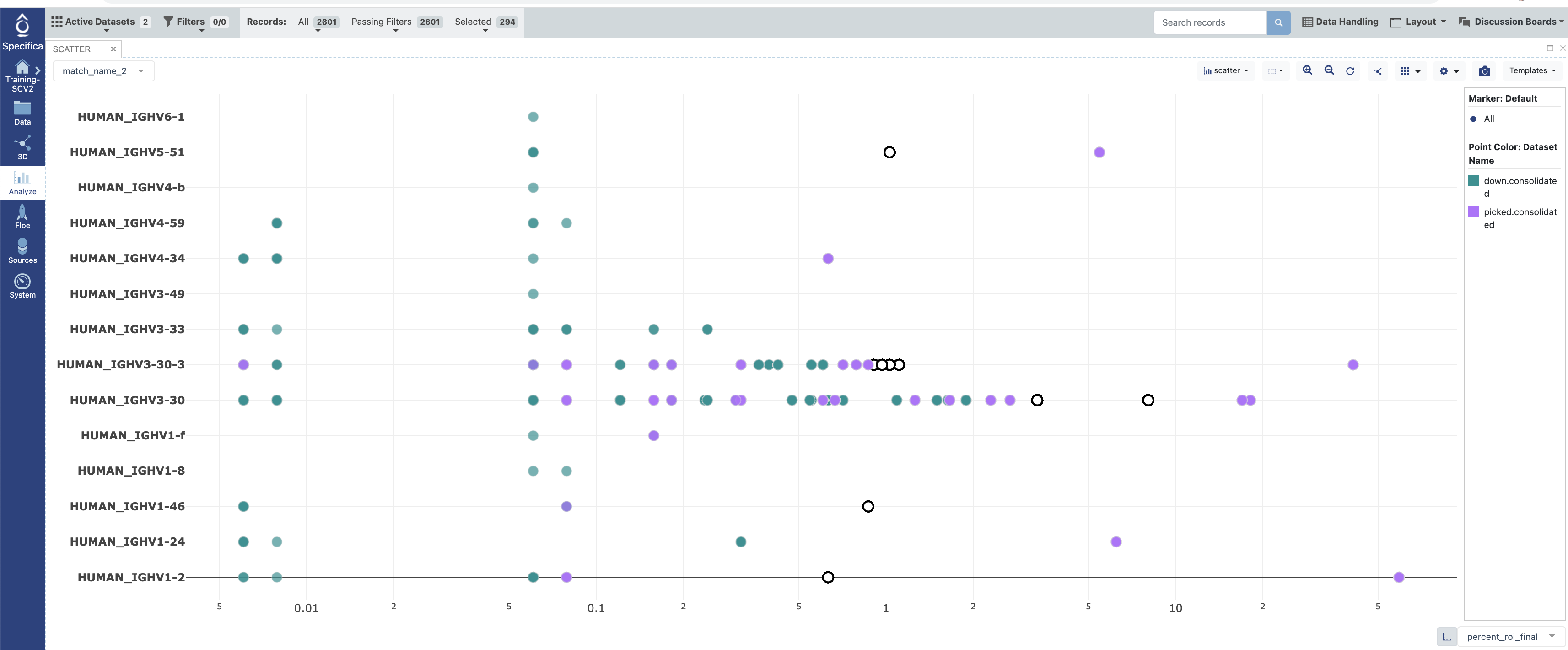
Open in the Analyze function and plot ‘percent_roi_final’ on the x-axis with the ‘match_name_2’ on the y-axis.
Under the settings for the plot color code the points by the “Dataset”.
Select populations found in the ‘down.consolidated’ dataset only, not the ‘picked.consolidated’.
Follow #12-14 under STEP 5 of Tutorial 3. We will ignore the well_id output as Sanger sequencing was not performed here.
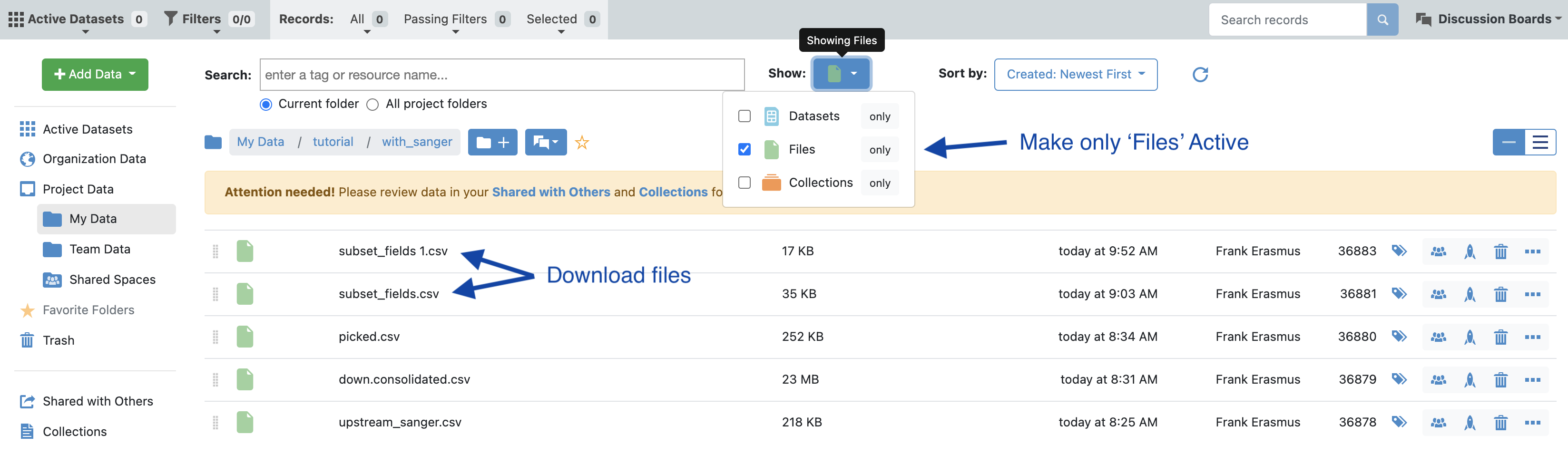
In the data directory, make files visible (see image) and download the csv files corresponding to the selected population. In this case we want the ‘subset_fields.csv’ which is derived from the additional clones selected in the Analyze tool as well as the ‘picked.csv’ which is the automated pipeline picked population.