Frequently Asked Questions
When should I change the memory or thread count?
Adjusting the memory and number of threads can be very important for running the most efficient QM calculations. The defaults of 14 GB and 8 CPUs were set to be conservative for our default methods and basis sets (HF-3c for geometry optimizations and B3LYP-D3MBJ/6-31G* for single point energies) for most drug-like molecules (up to 30 heavy atoms). If you are going to run calculations with a non-default method or basis set, we recommend performing smaller scale benchmark calculations to make sure your resource settings are sufficient. How to Run Benchmark Floes for Cost Estimation
How do I check the metrics (memory, CPU usage) in a QM calculation?
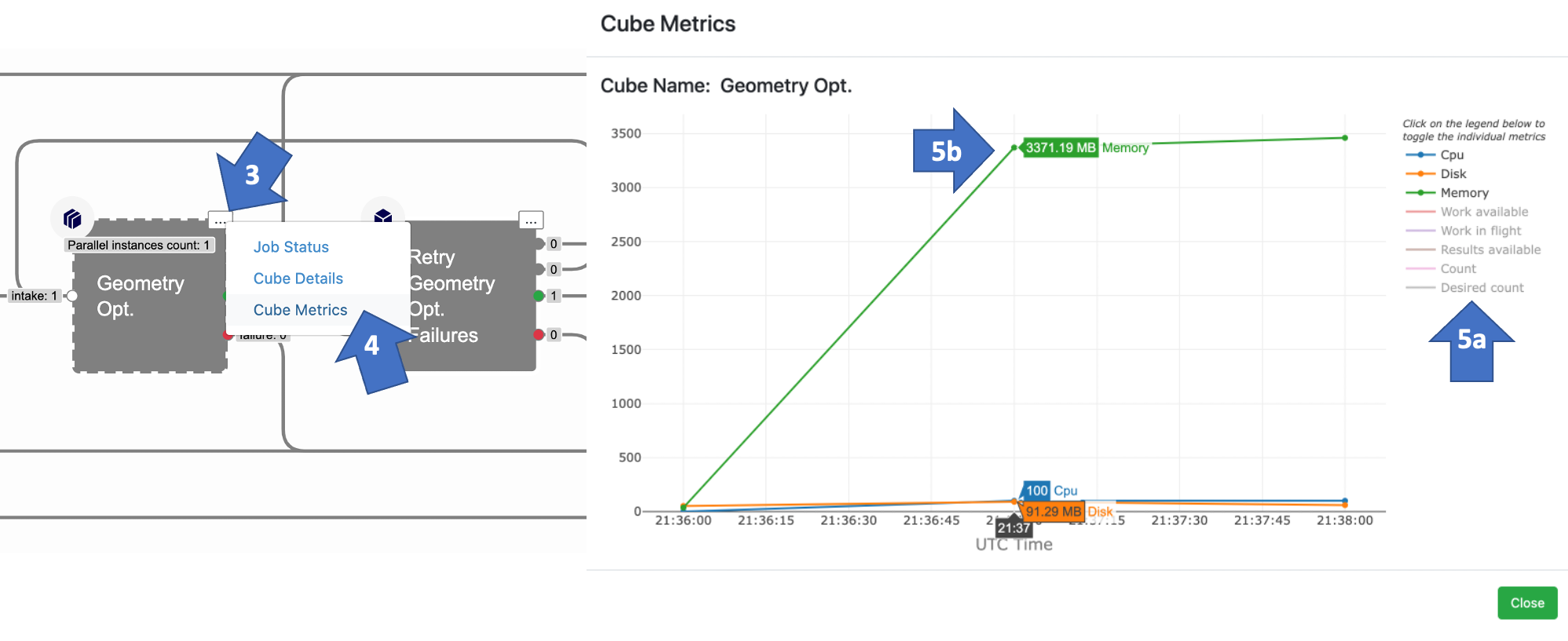
By default, the memory and CPU usage metrics are turned On for cube_Psi4PropertyCube and cube_Psi4GeomOptCube Cubes. That means you can see how much memory the cube actually used during a floe by following these steps:
Go to the Floe tab, click on Jobs, then select your running or finished floe.
Zoom in to the Geometry Opt or Single-Point Energy Cube in the floe.
Click on the ellipsis.
Select Cube Metrics.
A graph will appear showing how each cube metric changes over time.
You can deactivate a metric you do not want to see by clicking on its name in the top right.
When scrolling over the graph, the value for each line appears; this is helpful since the units for each metric are different.
How much will my floe cost?
This is a difficult question to answer. The computational cost of QM calculations are going to depend on both the size of your molecule and the method and basis set you have chosen for your calculation. Before changing the method or basis set or moving to much larger molecules (>30 heavy atoms), it is best to run benchmark calculations. How to Run Benchmark Floes for Cost Estimation.
In our floes, we have done our best to choose methods and basis sets that we believe have the best trade-off for computational cost and accuracy. We have chosen HF-3c as a default method (which incorporates the minix basis set) for most of our calculations. The very small basis set makes calculations with HF-3c very affordable, and in our experience the geometries of optimized conformers are sufficient for most uses. You will see in floes such as Psi4 QM Conformer Ensemble and Psi4 QM Local Minima Search where we want to compare relative conformer energies, we typically use a higher level of theory for the single point energy calculations.
What method and basis set should I use?
There is no universal right answer to this question. Before making changes, consider (1) if the default method and basis set are appropriate, (2) the type of calculation being performed, and (3) what elements and ions are included in your system.
1. Defaults
The defaults chosen for each floe have the goal of being both affordable and reasonably accurate for most neutral drug-like molecules. HF-3c – a corrected Hartree-Fock method from Sure and Grimme – is our default method (minix basis set) for most calculations [Sure-2013]. We have found HF-3c to be very reliable for geometry optimizations. However, this basis set is quite small, so if your molecule has atoms larger than argon, you may want to consider a larger basis set. It is common practice to follow up geometry optimizations with single point energy calculations at a higher level of theory. Our default for these calculations is the B3LYP-D3MBJ method with the 6-31G* basis set [Grimme-2011].
2. The type of calculation
Remember to think about the calculation being performed before changing the default method and basis set. If you are looking for a very accurate minimum geometry, then you may need a more sophisticated method and larger basis set. However, we have many floes, such as Psi4 QM Conformer Ensemble, where all dihedral torsions are constrained during optimization. A more expensive method and basis set are unlikely to change the optimized structure in that case, so the extra cost would be unjustified.
3. Heavy elements and ions
As mentioned above, the minix basis set used to fit HF-3c only supports elements up to argon. The 6-31G* basis set supports elements up to krypton. Molecules with larger atoms should use larger basis sets. In addition, most of our benchmarking has been performed with neutral molecules. If you are working with salts or other charged species, then you may also need larger basis sets. Any time you change the defaults, you should consider performing benchmark calculations to make sure the memory and thread count settings are sufficient, and you know how expensive large-scale jobs will cost.
Why are my calculations failing due to the presence of implicit hydrogens?
Implicit hydrogens can be added to a molecule in two ways: drawing a molecule in the Orion 2D Sketcher or from uploading an input file without bonding information.
In order to perform QM calculations on molecules drawn in the Sketcher, you will need to first generate coordinates for the molecule. This can be done with many floes in the OpenEye Classic Floes package, such as the OMEGA - 3D Conformer Ensemble Generation Floe. Alternatively, conformer ensemble and torsion scanning floes in this package will also generate conformers for those purposes.
When converting files without bonding information (i.e., XYZ files) to molecules on Orion, connectivity and bond order are perceived based on atom distances. For some molecules, this can lead to unexpected implicit hydrogens when bond orders are perceived at a lower order to satisfy the valence expected for each element. It is best practice to look at the uploaded datasets to check for these unnecessary hydrogens by checking the assigned bond order. Implicit hydrogens will appear in 2D images of the molecule, but not in the 3D page.
Implicit hydrogens make QM calculations impossible as they do not have 3D coordinates.
Therefore, when implicit hydrogens are found on
molecules in Gaussian Floes, the calculation will fail. If it is determined that these
implicit hydrogens were added by mistake, then make sure the Remove Implicit Hydrogens option is
set to On in the Advanced Section before running your floe. When this option is turned On,
implicit hydrogens are removed from the molecule before performing a QM calculation.
Alternatively, use one of the options above to generate coordinates for all of the atoms in your system.