Release Highlights 2026.1
Small Molecule Discovery Suite
ROCS X Floes
The new ROCS X Floe package is an add-on available in the Hit Identification Module of the Small Molecule Discovery Suite. It introduces a new generation of shape-based virtual screening workflows designed to handle libraries of trillions of compounds. ROCS X includes two fully automated floes for library generation with the Multi-Stage ROCS X Preparation Floe and search with the Multi-Stage ROCS X Search Floe.
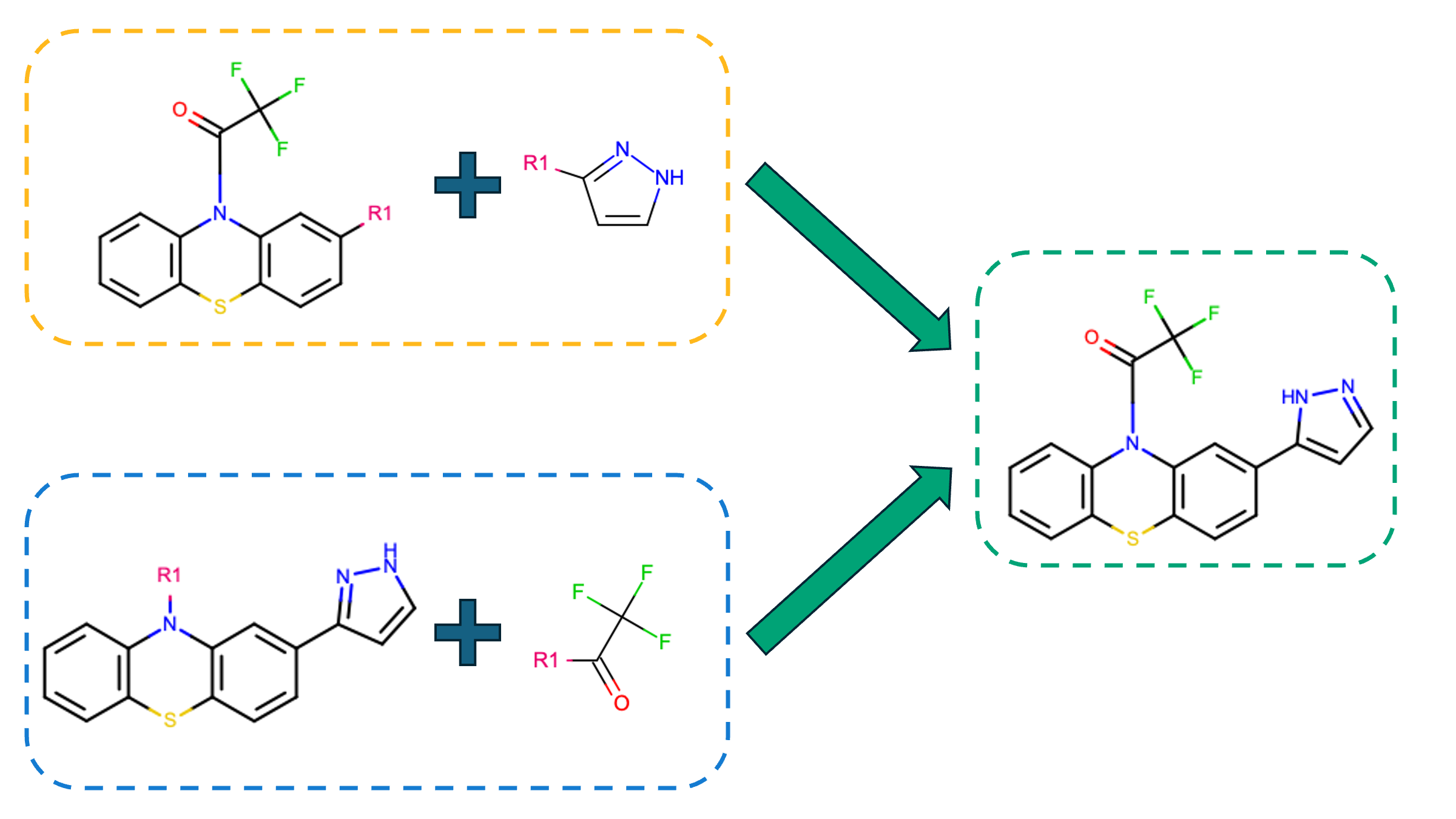
Figure 1. ROCS X efficiently searches in synthon space before using reaction definitions to make a final product. When multiple reactions (yellow box and blue box) result in the same product (in the green box), information for all possible reactions and reactants is provided in the final hit list.
ROCS X reaches these extreme scale libraries by using a combination of reagents and reactions to try to ensure synthetically accessible products. The libraries are generated in three stages: ingest building blocks and reactions to build a reaction and reagent database; convert building blocks into synthons which represent the chemistry of the fragments in the final product; and generate conformers to prepare the ROCS X library. Reagents are deduplicated at the synthon stage while maintaining information about all possible vendors. For reagents that include protecting groups, both protected and deprotected synthon states are automatically generated. Included with this release are prebuilt libraries of up to 11 trillion compounds created with building blocks sourced from leading vendors, including Enamine, MolPort, and Mcule.
Searching in ROCS X can also be performed in an automated floe with three stages. First, an AI model in the form of a multi-arm Bayesian bandit is initialized from an input query and a sample of products in the provided ROCS X library or a predefined hit list. Second, the model is used to sample products from the library based on the shape of each synthon. When necessary, duplicate products are removed while maintaining information about the different combinations of reagents and reactions that lead to those duplicates. Third, the final hit list is clustered based on scaffolds and 2D fingerprints, as well as providing a diverse representative subset of molecules suitable for downstream applications.
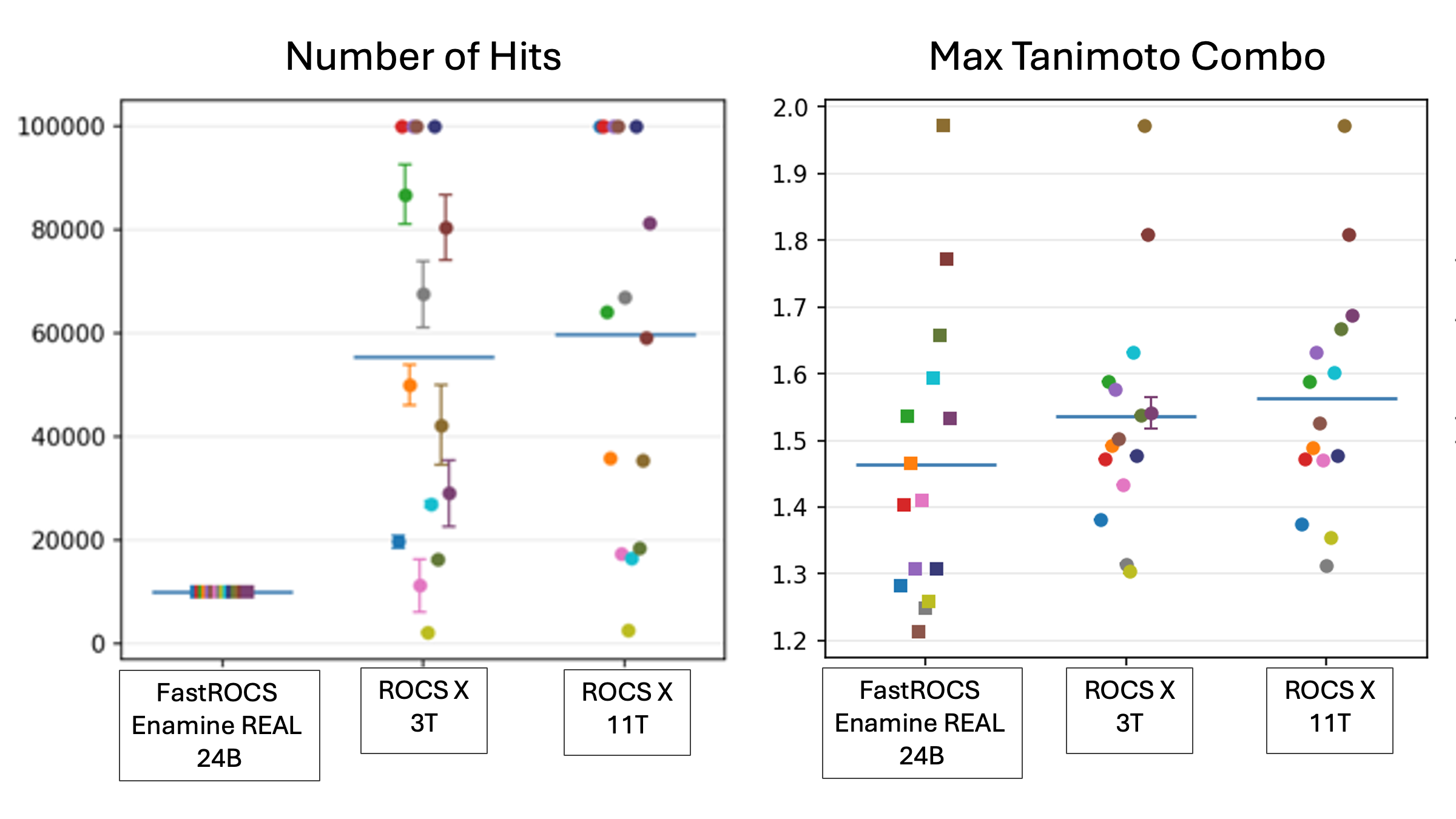
Figure 2. These plots show results for multiple runs on color-coded queries comparing ROCS X to a fully enumerated FastROCS search of an Enamine collection. ROCS X results in larger hit lists with higher similarities. The ROCS X hit lists (left plot) include all molecules with a Tanimoto combo greater than the minimum found in the 10,000 top hits from FastROCS.
By searching libraries of trillions of compounds, more hits with greater shape similarity can be found in comparison to searching with pre-enumerated commercial libraries. These floes leverage AI and FastROCS to search synthetically accessible libraries at this extreme scale. As a virtual screening tool, ROCS X has had a proven impact on diverse discovery programs across therapeutic areas.
Large Scale Floes
Several improvements have been made to the Large Scale Floes package to improve the flexibility of these floes. The Batch FastROCS and FastROCS Plus Floes have been updated to support more predefined color force fields or fully customized color force fields. The Batch FastROCS Floe now supports any ROCS query type, including shape grids without molecules.