Frequently Asked Questions
When should hardware requirements be changed?
The memory, disk space, and number of threads parameters can be very important for running the most efficient QM calculations. The defaults of 14 GB memory, 25 GB disk space, and 8 CPUs were set to be conservative for our default methods and basis sets (HF/6-31G for geometry optimizations and B3LYP/6-31G* for single-point energies) for most drug-like molecules (up to 50 heavy atoms). If you are going to run calculations with a non-default method or basis set, we recommend performing smaller scale benchmark calculations to make sure your resource settings are sufficient.
How should hardware metrics be checked in QM calculations?
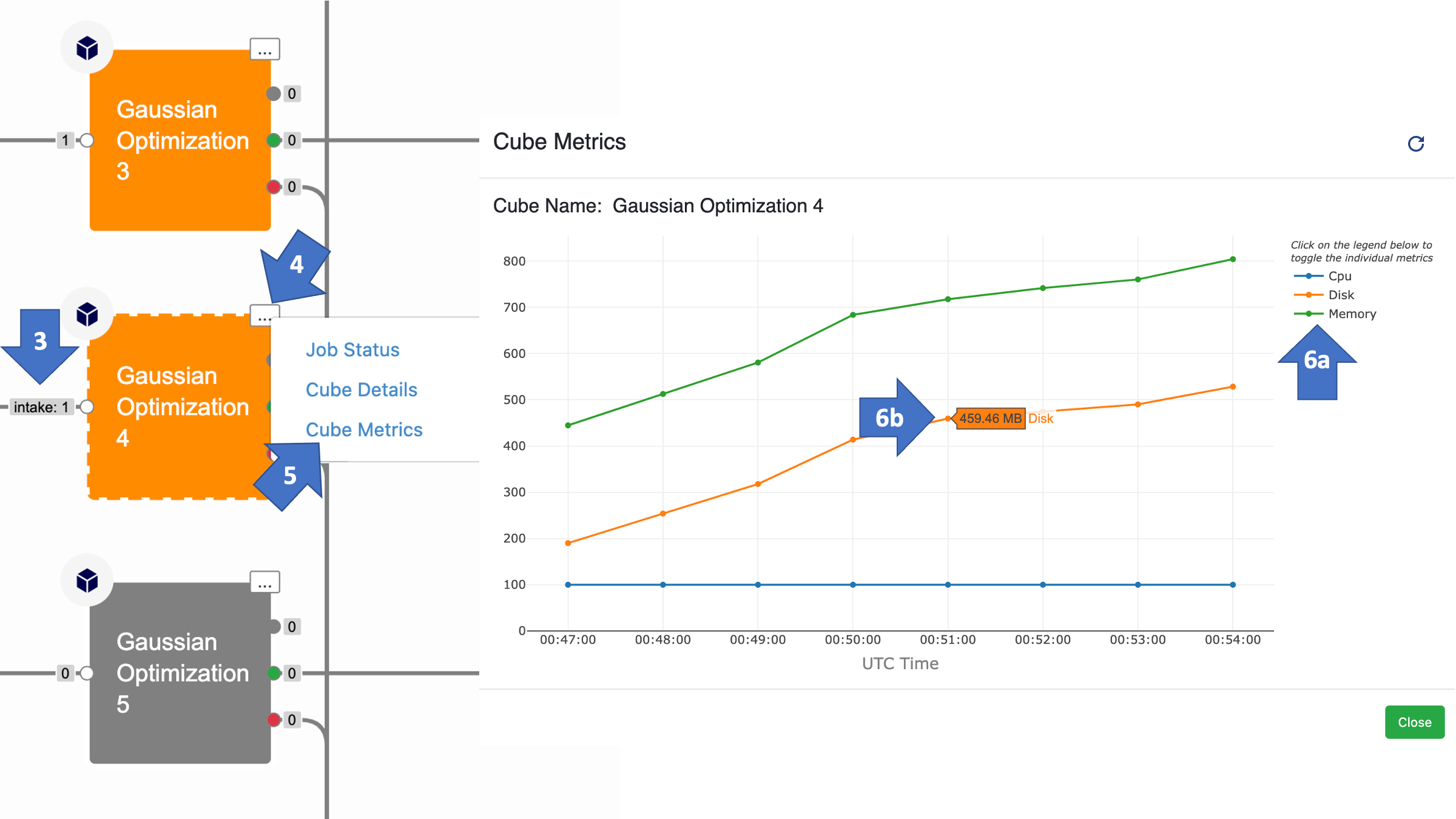
By default, the memory, disk space, and CPU usage metrics are turned on for all three cubes which use Gaussian Software (Run Gaussian Input Directories, Gaussian Single Point Energy, and Gaussian Geometry Optimization Cube). That means you can see how much memory and disk space the cube actually used during a floe by following these steps:
Go to the Floe tab, click on Jobs, then select your running or finished floe.
Zoom in on the calculation cube(s) in the floe.
Make sure to select a cube that had one or more records pass through it.
Click on the ellipsis.
Select “Cube Metrics.”
A graph will appear showing how each cube metric changes over time.
You can deactivate a metric you do not want to see by clicking on its name in the top right.
When scrolling over the graph, the value for each line appears. This is helpful since the units for each metric are different. The memory and disk space are shown in MB and the CPU is a percentage of requested CPUs (i.e., 100 means 8 CPUs out of the 8 requested are being used).
How much will a Floe cost?
This is a difficult question to answer. The computational cost of QM calculations is going to depend on both the size of your molecule and the method and basis set you have chosen for your calculation. When changing the method or basis set or shifting to much larger molecules (>50 heavy atoms), it is best to run a small benchmark calculation while monitoring the cube metrics on Orion.
What method and basis set should be used?
There is no universal right answer to this question. Before making changes, consider whether the default method and basis set are appropriate, what elements and ions are included in your system, and the type of calculation being performed.
The defaults chosen for each floe have the goal of being affordable and reasonably accurate for most neutral drug-like molecules. HF/6-31G and B3LYP/6-31G* are the defaults for geometry optimizations and single-point energies. These are both relatively small basis sets. If your calculations include charged species or elements larger than krypton, a larger basis set may be required. Any time you change the defaults, make sure to monitor the cube metrics to make sure the memory, disk space, and thread count settings are sufficient. Consult the Gaussian documentation for more information on DFT methods and basis sets.
Remember to think about the calculation being performed before changing the default method and basis set. If you need minimized geometries at a high level of theory, it can be more efficient to start with a lower level of theory. For example, if starting with conformers from Omega, which uses a force field to set bond lengths and angles, a geometry optimization at the default level of theory should be performed first. Then perform a second optimization at the higher level of theory.
How can Gaussian log files be used to understand failures?
There are a variety of errors that can cause Gaussian input files to fail. The Gaussian QM Run Input Files Failed Calculation tutorial demonstrates the two ways calculations can fail: (1) an input file is rejected by Orion and (2) the Gaussian calculation fails.
In the first category, failures are relatively easy to understand. The hardware requirements in the Gaussian input file must agree with the resources requested on Orion. In this case, the Failure Report should provide enough information for how to change an input file to make it successful. It is best practice to not specify thread count, memory, or disk space in an input file; instead use the Orion Hardware Requirements to meet your needs.
Understanding Gaussian calculation failures can be more complex. The best option is to download the resulting archive files from a calculation. There is a subdirectory for each calculation. The log files include details of where in the calculation Gaussian failed. For help understanding these log files, consult the Gaussian documentation or see Gaussian’s instructions for technical support. In addition, there are many open source resources about common Gaussian errors, including those in the Compute Canada Documentation and Zhe Wang’s Blog.
Why are there multiple serial cubes for running input files?
In the Gaussian QM Run Input Files floe there are many copies of the cubes that performs the Gaussian calculation. Orion has a variety of different cube types. Parallel cubes request resources on demand from AWS based on the number of inputs. While this should be ideal for scaling up any calculation, parallel cubes also have some important limitations. The primary concern for Gaussian calculations is that parallel cubes have a strict time limit, and calculations that exceed that time limit are canceled. Given the flexibility in calculations performed with Gaussian input files this time limit needs to be avoided. When running many input files for short calculations, the floe Gaussian QM Parallel Run Input Files can be used instead.
How do you create datasets on Orion?
Datasets on Orion can be created by uploading any standard molecule file (mol2, sdf, xyz, etc.). These files will automatically be converted to datasets with 1 record/molecule. If you upload file types which have coordinates, but no connectivity information (such as XYZ), check your uploaded dataset and see the information about implicit hydrogens below.
Datasets can also be created with SMILES files or by drawing a molecule in the Orion 2D Sketcher. This is a simple option that is best paired with the Gaussian QM Conformer Ensemble or the Gaussian QM UI Torsion Scan Floes where coordinates will be generated for your input files.
Why are my calculations failing due to the presence of implicit hydrogens?
Implicit hydrogens can be added to a molecule in two ways: drawing a molecule in the Orion 2D Sketcher or uploading an input file without bonding information.
In order to perform QM calculations on molecules drawn in the Sketcher, you will need to first generate coordinates for the molecule. If you have access to the Small Molecule Discovery Suite, this could be done with the OMEGA - 3D Conformer Ensemble Generation floe*. Alternatively, a full conformer ensemble can be generated with the Gaussian QM Conformer Ensemble floe in this package.
When converting files without bonding information (such as XYZ files) to molecules on Orion, connectivity and bond order are perceived based on atom distances. For some molecules, this can lead to unexpected implicit hydrogens when bond orders are perceived at a lower order to satisfy the valence expected for each element. It is best practice to look at the uploaded datasets to check for these unnecessary hydrogens by checking the assigned bond order. Implicit hydrogens will appear in 2D images of the molecule, but not in the 3D page.
Implicit hydrogens make QM calculations impossible as they do not have 3D coordinates.
Therefore, when implicit hydrogens are found on
molecules in Gaussian floes, the calculation will fail. If it is determined that these
implicit hydrogens were added by mistake, then make sure the Remove Implicit Hydrogens option is
set to On in the Advanced Section before running your floe. When this option is turned On
implicit hydrogens are removed from the molecule before performing a QM calculation.
Alternatively, use one of the options above to generate coordinates for all of the atoms in your system.
How do you rerun a Gaussian calculation from a checkpoint file?
Checkpoint files are only generated if they are specified in the
Gaussian input file with link0 entry %chk.
If you have completed a calculation with a checkpoint file, then
that file can be used as part of the input for a future calculation.
The only requirement is that the checkpoint file is in the same
subdirectory before archiving it into a tar or zip file for the
calculation. For more information about checkpoint files, refer to the
Gaussian documentation for input files.
Can GPUs be used with Gaussian on Orion?
The short answer: no.
This decision was made by considering the cost increase and availability of GPUs on Orion compared with the expected time savings. For now, this trade-off does not seem to be worthwhile. This may change in future releases. If you need GPUs for your Gaussian calculations, please contact support@eyesopen.com.
How should files for the Run Gaussian Input File Floe be created and organized?
The Gaussian QM Run Input Files Floe looks for Gaussian input files with .com or .gjf extensions. This floe can also take tar or zip files with multiple input directories. The floe can also accept multiple files of any type as input. Tar files with no compression or zip, gzip, bzip2, or lzma compression can be processed. In case of multiple .com or .gjf Gaussian files, you can keep all the files in the main directory or any organization of subdirectories before archiving.
When there are multiple input files in the same directory, a new subdirectory will be created for each file with the same name as the file. If duplicate files or directories are found when parsing all input to the floe, an integer will be added to the end of the subdirectory name in order to reduce the risk of lost data. However, this could cause confusion when analyzing the output, so it is best practice to make sure all Gaussian input files have unique names, even if multiple archives are passed to the same floe.
The output from all calculations in one job will be saved to a single output file. The output files must be tar files with or without compression. The type of tar file is determined by the extension. If an unsupported extension is provided, a tar file without compression will be created (that is, if the output file is named Gaussian_output.zip, Gaussian_output.tar is created instead).
Why did the Gaussian input file change after running on Orion?
When running Gaussian files on Orion, those files are validated in a number of ways, some of which may result in the file being changed. Any references to memory, thread counts, or disk space in the input file are compared to the hardware requirements requested for the floe. If more memory, disk space, or threads are specified in the input file, the calculation will fail.
There is only one situation where an input file could be changed. If an input file specifies a path to any of the Gaussian output files, then those paths are reset. There is limited write access for cubes on Orion, so output file paths are reset to be written to the directory where the calculation was run. This means those files will be included in the output.
What should you know before running a large calculation with a Gaussian input file?
The ability to run a Gaussian calculation with any input file allows for flexible calculations. However, the default hardware requirements for this floe were set to the same levels as the Single Point Energy and Geometry Optimization Floes. That is, they should be sufficient for DFT calculations on most drug molecules (<50 heavy atoms) with relatively small basis sets (such as B3LYP/6-31G*). When running calculations with larger molecules or higher levels of theory, keep a close eye on the metrics on Orion..
It is also best practice to always save your
checkpoint file
by adding the line %chk=[filename].chk to your input file.
These files save the state of your system and are useful for
restarting a calculation
when necessary.