How to Generate Conformers with a Custom Torsion Library
Disclaimer: Running both floes in this tutorial will only cost about $2 with some variation based on current AWS pricing. Remember that changes to QM methods and basis sets or molecule size could significantly change costs. If you wish to run this tutorial with a much larger molecule than the one provided, or with a non-default method and basis set, you may wish to review How to Run Benchmark Floes for Cost Estimation.
This tutorial uses the following floes:
Create an Input Dataset
You can copy a SMILES from anywhere and paste it into the Orion Sketcher.
The fragmentation and torsion scanning floe can take any molecule from the
Sketcher or an existing dataset. For this tutorial we use a molecule from the
Crystallography Open Database with
SMILES COC(=O)c1cc([nH]n1)c2ccccc2 and COD identification
number 2216060.
For this tutorial, you can paste the SMILES into the Orion Sketcher or
follow along with your own molecule.
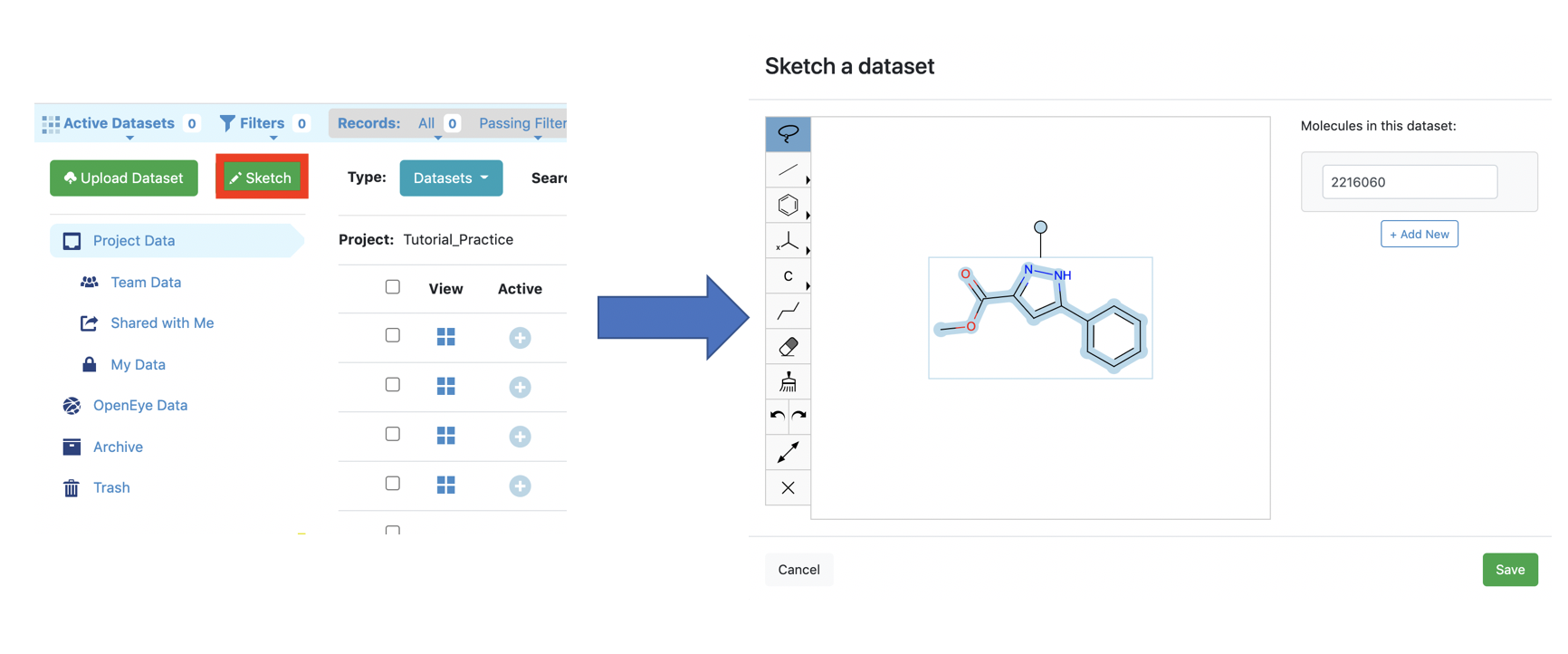
From the Orion Data page, find the Sketcher and paste the SMILES or draw your own molecule. Consider that floe costs could change dramatically with molecule size and number of rotatable bonds.
Run the Fragmentation and Torsion Scanning Floe
Locate the Psi4 QM Fragmentation and Torsion Scanning in the category path Product-based / Quantum Mechanics / Psi4.
Parameters to change
Select Input Dataset
For this tutorial we will use default parameters. Therefore, all you need to do is to select your input dataset and rename your outputs, if desired.
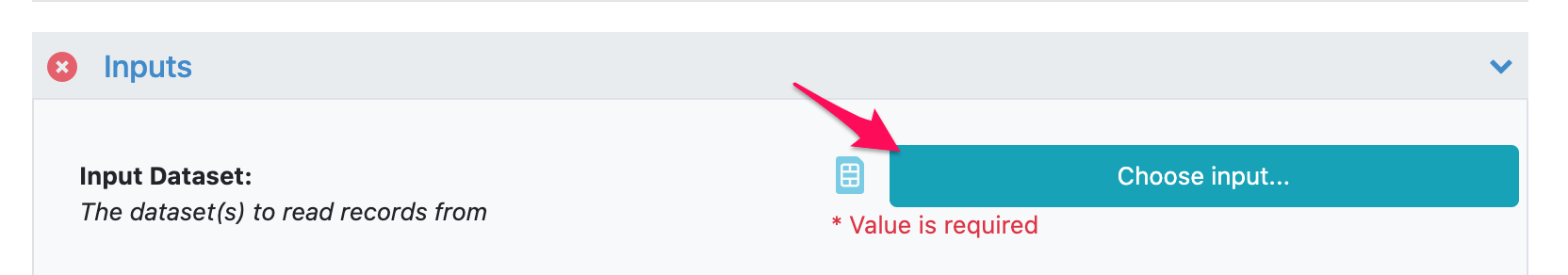
Select “Choose input…” and find the dataset you created above, or use the sketch tab at the top right to draw a new molecule.
Specify Output Datasets

Default output dataset names are always available. One way to keep track
of floe output is to add a prefix to these so you know which job they came from.
In this case, the prefix psi4_torsion_tutorial was added to the beginning of
each of the outputs generated by this floe.
Torsion Rules Output: This option saves the input molecule with the custom torsion rules from the fragment scans as a new field on the record.
Fragment Output: In this case, it indicates the fragments generated around each rotatable bond in the input molecule. Each molecule in the output has a conformer record for each angle in the torsion scan, about 73 in total.
Failure Output: This only appears if there are any failed calculations. It is important to note that during QM torsion scans, there are sometimes failures for very high energy torsions which fail to converge during optimization. Generally, we do not worry about these failures. A report will be generated at the end summarizing any failures, which we will investigate.
Fragmentation Report Title: This is a Floe Report (html file) with the torsion scan results.
Parameters to leave unchanged for tutorial
Torsion Increment: This is the separation between points in the torsion scan in degrees. It is not recommended to set this value above 10 degrees when you wish to use the custom torsion library.
Psi4 Calculation Parameters: This collapsed parameter group includes the parameters for changing the method, basis set, memory, and number of CPUs used by the QM optimization. For each torsion increment, in each fragment, a geometry optimization is performed constraining that torsion while allowing all other degrees of freedom to relax. We have chosen HF-3c (minix basis set) as a default method for this protocol because it gives a reliable sense of the energy landscape for drug-like molecules. The memory and CPU defaults should be more than sufficient for most molecules under 30 heavy atoms. If you are working with a much larger molecule, or a different method or basis set, you may need to perform some benchmark calculations first; see Benchmark Torsion Scanning Floes.
Understanding the Torsion Scan Floe Report
Now that the floe has finished, you can see the torsion scanning results in the floe report. To find the report:
Navigate to the completed job from the floes page.
Click on the “Floe Report” tab above the floe diagram.
Click on the name of the report you specified above; in this example we used “psi4_torsion_tutorial_Fragmentation_Report.” Note that if you did not change the default output, it will just be called “Fragmentation Report.”
Optionally, click the square with an arrow to open the report in a new tab.
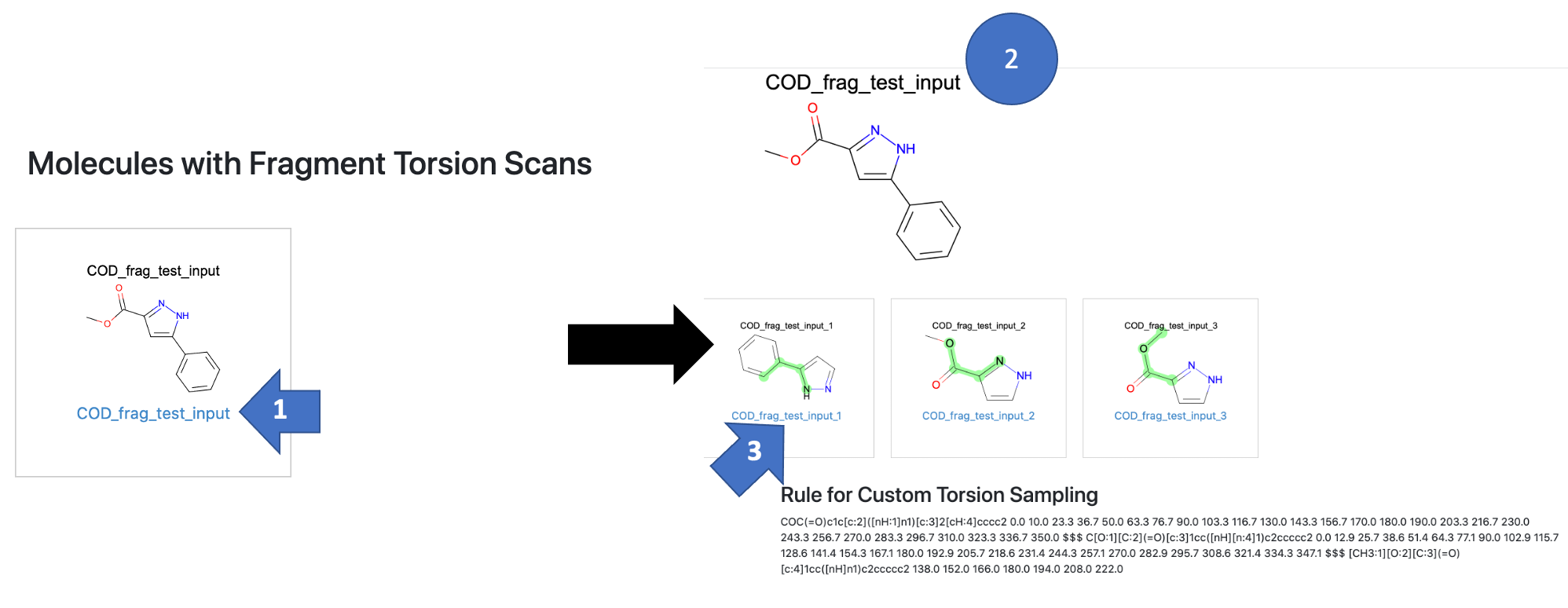
In the report, you will navigate by molecule. The first page is a grid of all molecules used as inputs to the floe. There was only one for this tutorial. To examine the results:
Click on the molecule whose fragments you want to see.
This takes you to a page showing the fragments created around rotatable bonds in this molecule (3 fragments in this example). The sampling rules generated for the molecule are also shown.
These sampling rules can be difficult to read as they are complex SMARTS patterns. They are stored in your output dataset so that they can be used as an input in the conformer-generating floes. If you want to understand the rules, they are written as a SMARTS pattern for the torsion followed by the angles to be sampled. The rules for different torsions are separated by $$$.
Click on any fragment to see the torsion scan for that bond.
On each fragment page, the torsions are shown:
On the fragment, the molecule used for the the torsion scan.
The same torsion highlighted on the full molecule for context.
The actual torsion scan, showing the relative QM energy at each angle increment highlighting the angles chosen for the custom torsion rule.
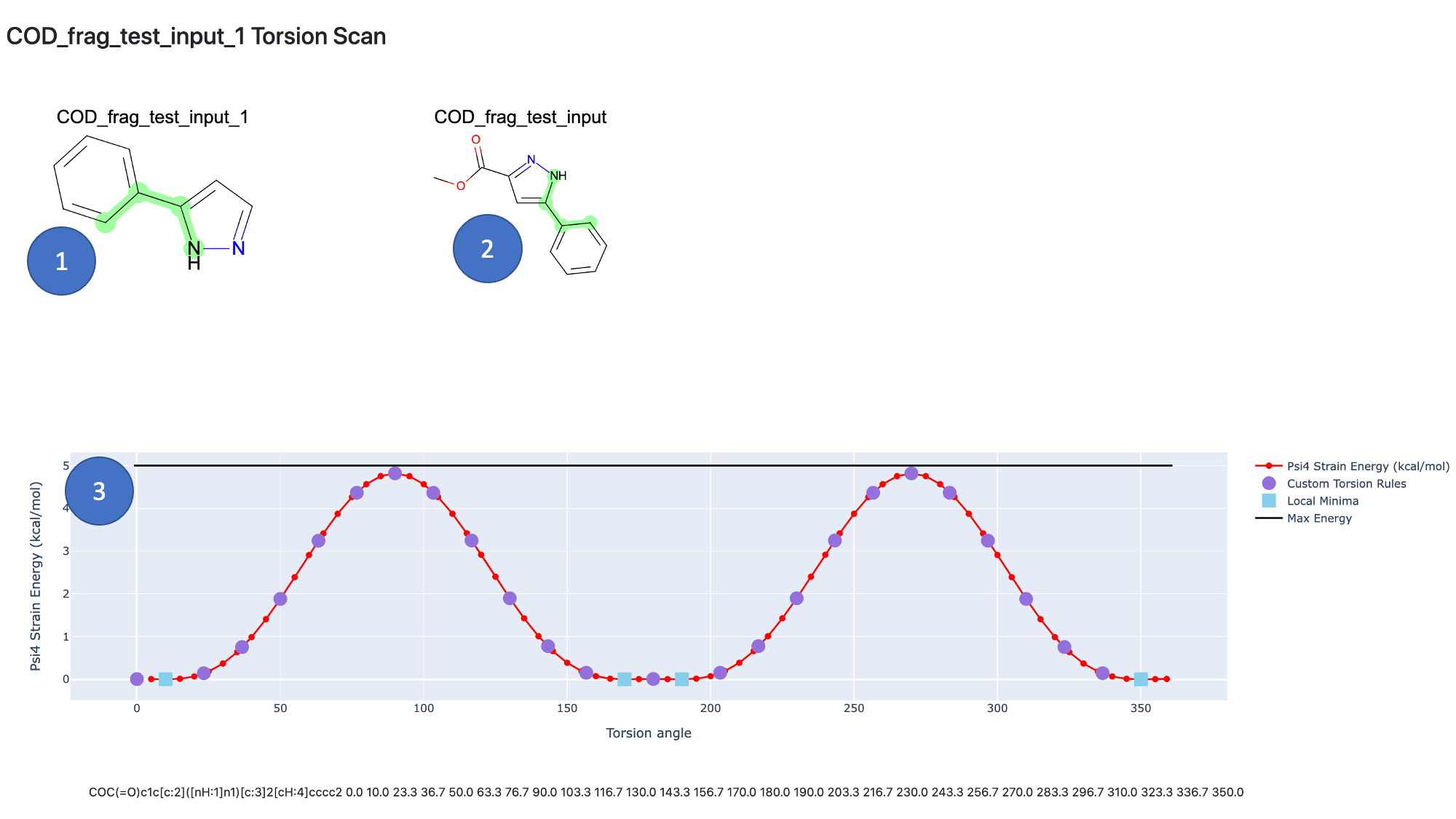
The browser back button can be used to navigate back to the previous pages.
Generate Conformers with Your Custom Torsion Rules
Locate the Psi4 QM Conformer Ensemble in the category path Product-based / Quantum Mechanics / Psi4.
The conformers generated in this floe are optimized while constraining torsions around all rotatable bonds. This allows for reasonable energies at the specified level of theory for a very diverse set of conformers. If you would prefer full conformer optimizations, the Psi4 QM Local Minima Search includes unconstrained geometry optimizations, and the parameters are nearly identical.
Parameters to change
Select Input Dataset
In order to use the torsion library created in the last floe, you need to use the
Torsion Rules Output. The default name for that output is``torsion_rule_output``
which corresponds to psi4_torsion_tutorial_torsion_rule_output if you used the outputs specified in
this tutorial.
Specify Output Datasets
As above, we will prefix each output with psi4_torsion_tutorial.
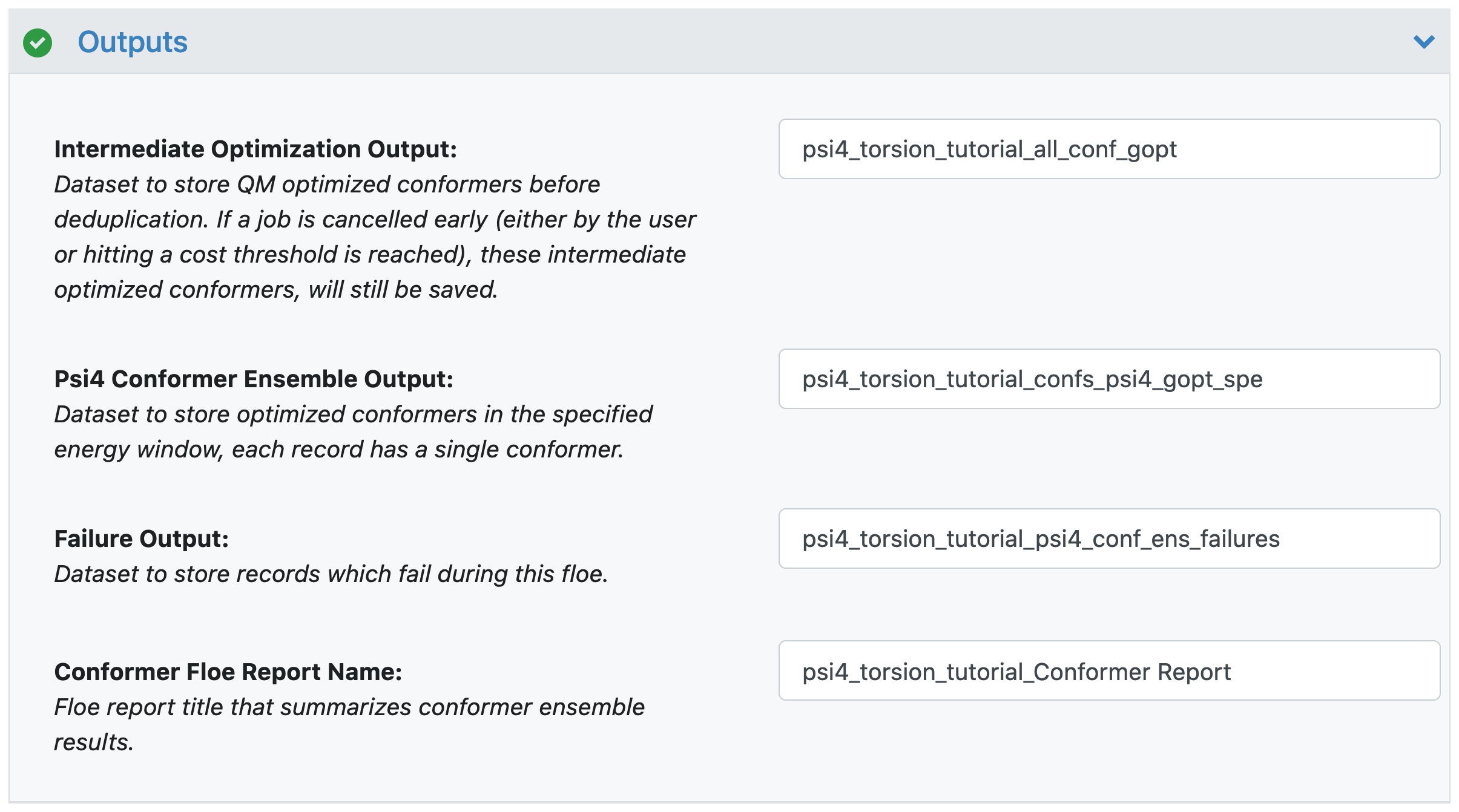
Intermediate Optimization Output: These records contain molecules which have completed torsion constrained optimizations, but the single point energy has not been completed. We save these output as an intermediate. If you need to quit the floe early or something goes wrong during the single point energy calculations, this intermediate output allows you to save the results from the optimization calculations.
Psi4 Conformer Ensemble Output: This is the final conformer ensemble with conformers which fall within your specified energy window.
Failure Output: These are records for calculations that failed during the floe. This includes conformers that are too high in energy to be included in the successful ensemble. Another cause for failures is that constrained optimizations for high energy conformers do not converge. During conformer generation, we use a force field in OpenEye Omega TK to evaluate conformer energies, but allow for fairly high energy conformers to guarantee sufficient sampling. It is not unusual for the QM optimization of these conformers to fail.
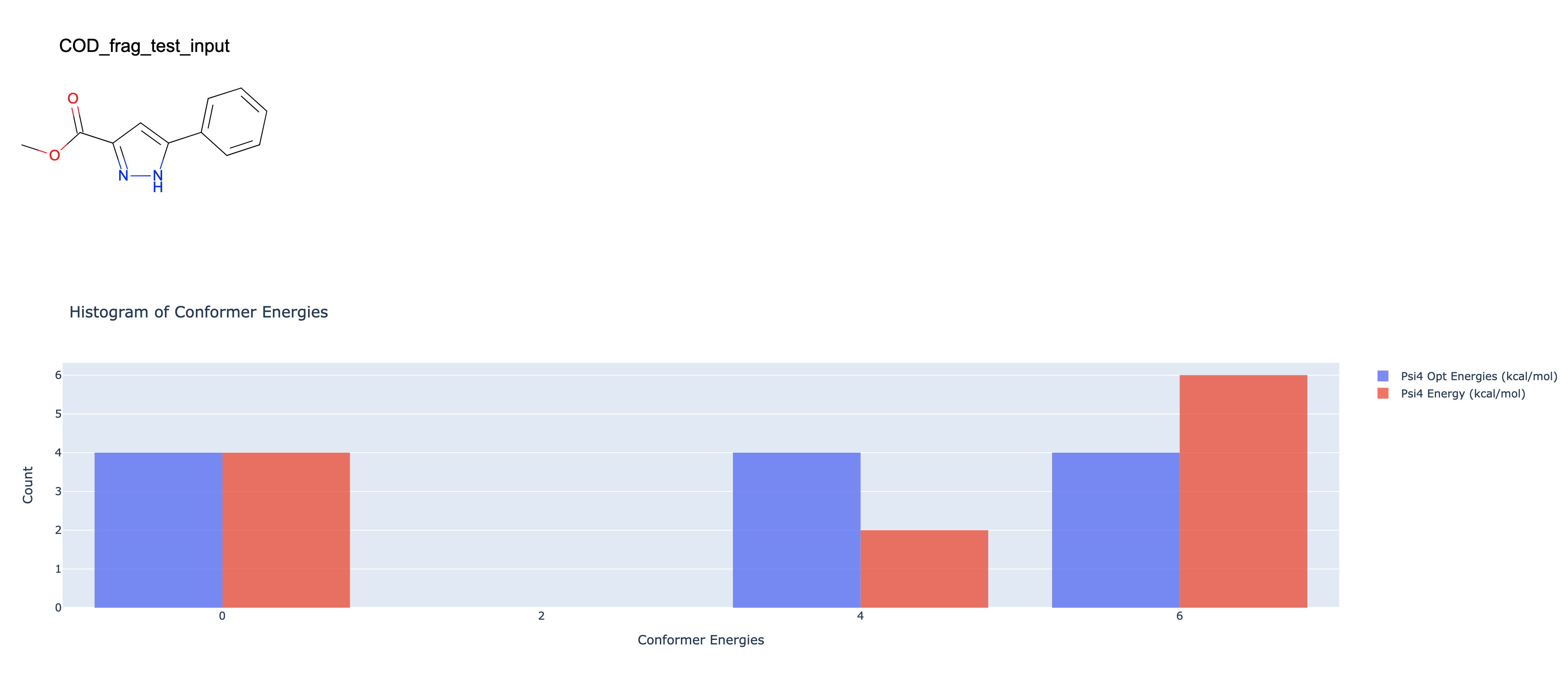
Conformer Floe Report Name: This is a floe report which summarizes the conformer ensemble results, including a distribution of energies in the final ensemble.
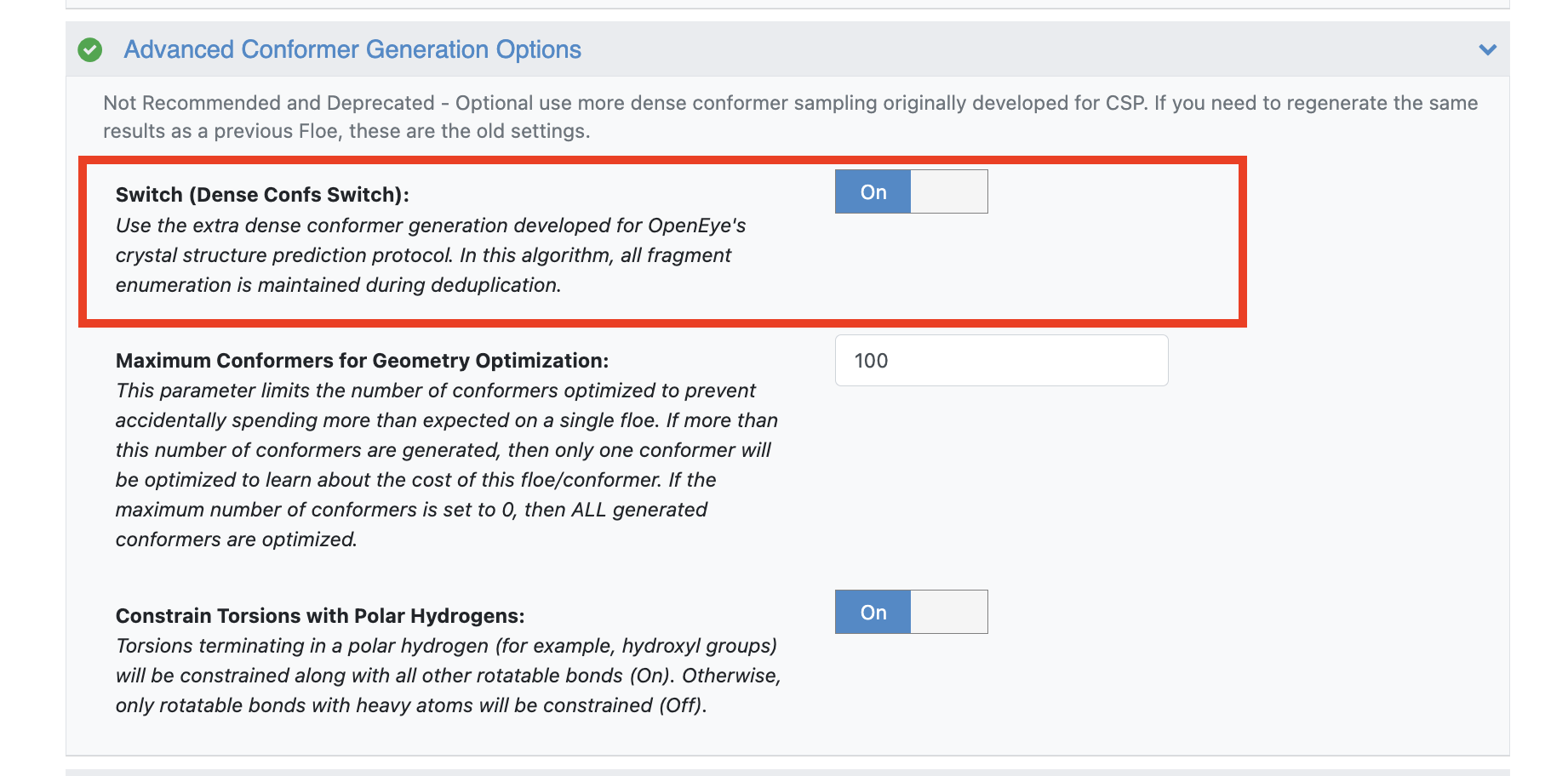
Use Dense Conformer Ensemble
Expand the “Advanced Conformer Generation Options” section to see all options for conformer generation. In order to use the custom torsion library, the Dense Conf Switch must be turned on.
Parameters to leave unchanged for tutorial
- Conformer Parameters:
RMSD Threshold for Conformer Generation: Dense conformer generation is performed with this as the maximum RMSD threshold between conformers.
Maximum Conformers for Geometry Optimization: This parameter limits the number of geometry optimizations performed. If the number of conformers generated is greater than this number, then only one conformer is optimized. That single conformer allows you to predict how expensive the full floe would be. See Benchmark Conformer Generating Floes for more details.
Psi4 Energy Window (kcal/mol): Only conformers within this energy threshold of the lowest energy conformer will be included in the final ensemble.
Constrain Torsions with Polar Hydrogens: When On, torsions terminating in a polar hydrogen (such as hydroxyls) are constrained with other torsions around rotatable bonds.
- Psi4 Calculation Parameters:
The biggest concern here is an accuracy/cost trade off; if you are going to change these settings, you may want to run a benchmark calculation with a strict limit on the number of geometry optimizations performed. Benchmark Conformer Generating Floes
Psi4 Hamiltonian (Geometry Optimization) and Psi4 Basis Set (Geometry Optimization): Remember that during the geometry optimization, all torsions are constrained. This allows the bonds and angles to relax, reducing the noise in your final energy while maintaining the diversity in the conformers generated. In our experience, for atoms up to argon, HF-3c does a good job of getting these small optimizations correct. If you are going to change the method and basis set for the optimizations, keep the constraints in mind considering the cost/accuracy trade off.
Psi4 Hamiltonian (Single Point Energy) and Psi4 Basis Set (Single Point Energy): If you are looking for a higher level of theory for the relative energy of your conformers, this is where you might want to make changes.
Psi4 Memory and Psi4 #Threads: These should be more than sufficient for our default method and basis sets with molecules up to 40 heavy atoms. If you have extremely large molecules or are going to change the method or basis set, you may want to perform a benchmark calculation and check the relevant cube metrics (please see: How do I check the metrics (memory, CPU usage) in a QM calculation?).
Understanding the Conformer Ensemble Floe Report
The floe report for the Conformer Ensemble can be found in the same way as above, under the Floe Report tab.