Gaussian QM Geometry Optimization Tutorial
Running this floe with the provided input dataset will cost around $0.45. Navigate through the categories Product-based / Quantum Mechanics / Gaussian to find this floe. Then locate or search for Gaussian QM Geometry Optimization.
The input for this floe (hydroxymethyl-benzoic-acid) can be found in the Gaussian QM Floes folder of the Tutorial Data on Orion. This floe requires 3D input molecule(s) or conformers to optimize (help generating datasets). In this tutorial, we will run the floe twice with different parameters and compare the results.
For the first calculation in this tutorial, we will perform a geometry optimization in gas phase with HF/6-31G*. In this calculation, change the basis set from 6-31G to 6-31G* and add gas to all of the output parameters to help distinguish the two results in this tutorial.
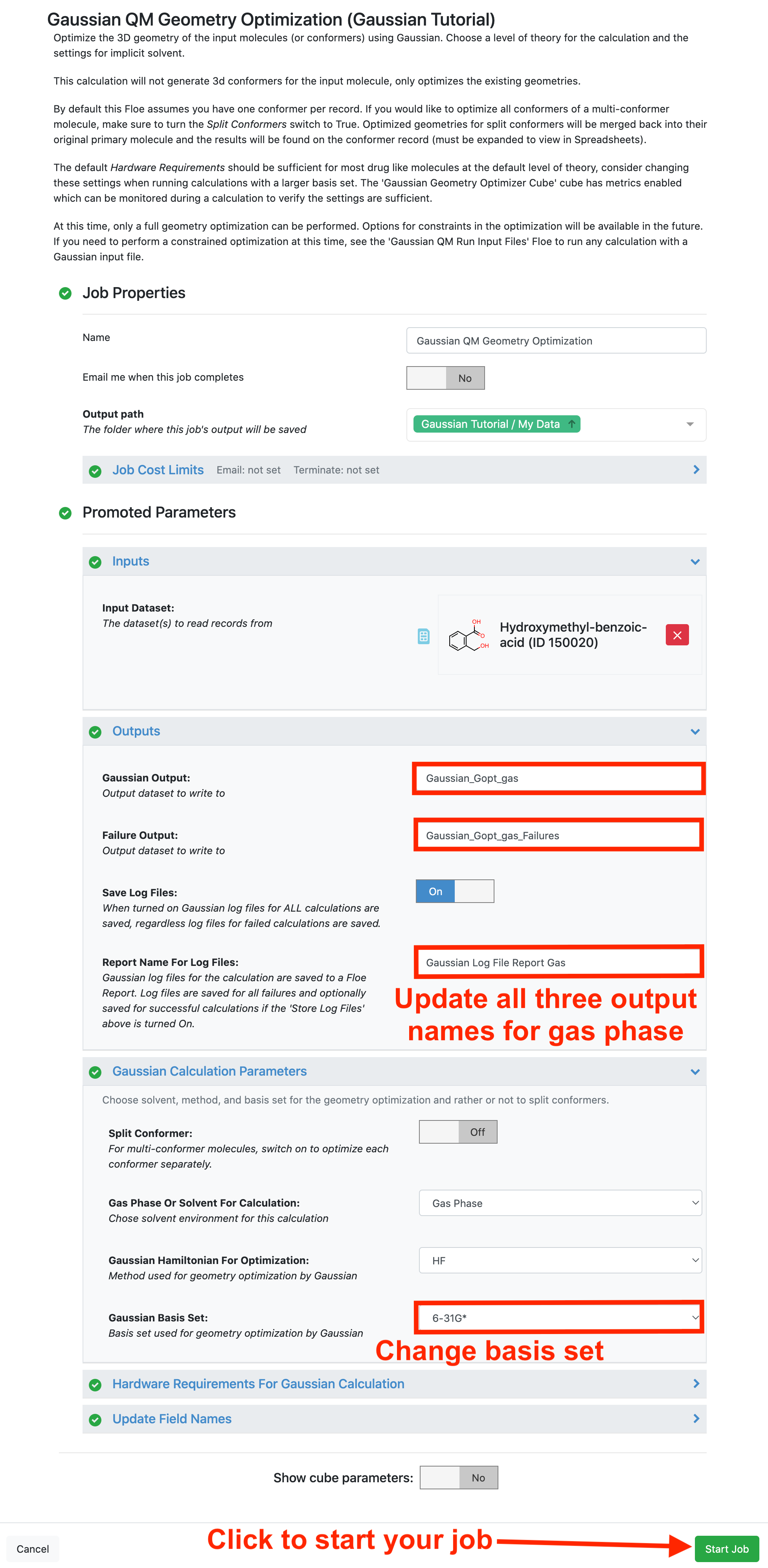
The default Hardware Requirements should be more than sufficient for drug-like molecules at the default level of theory and similarly sized basis sets. Consider changing these settings when running calculations with much larger basis sets. The metrics can be monitored on the Gaussian Geometry Optimizer Cubes to verify the settings are sufficient. There are multiple copies of this serial cube, so make sure to check the cube that the records are passing through.
Navigate to the Jobs tab and select this running floe to monitor the status of the calculation. At the bottom of the left menu is a “go” button that will bring up the job form with the same parameters.
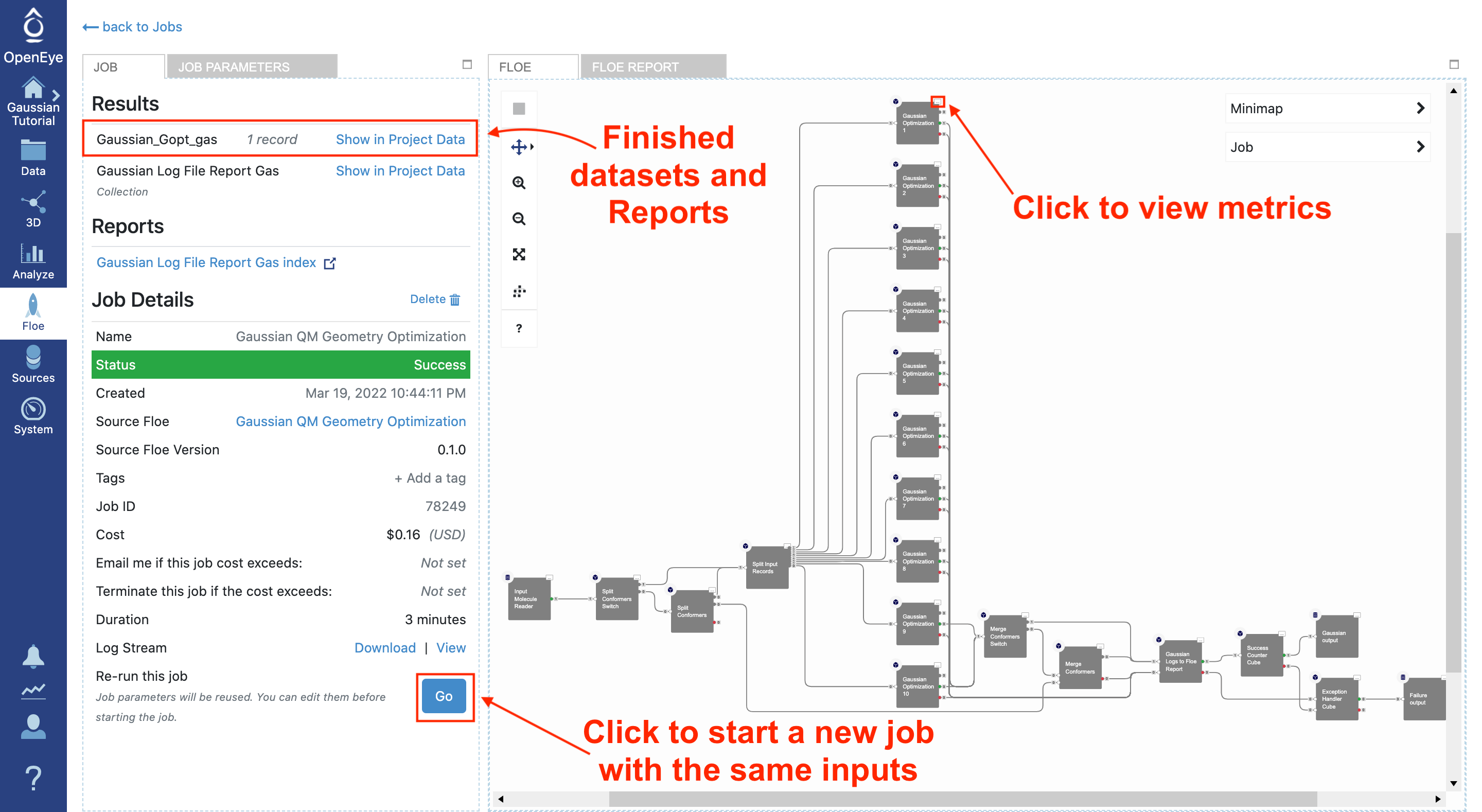
The job form will appear with the same input dataset and parameters. This time set the Gas Phase Or Solvent For Calculation parameter to “Water.” To distinguish these results from the gas phase calculation, add water to the output dataset names.

Note
At this time, only a full geometry optimization can be performed in this floe. Constraints will be added in future releases.
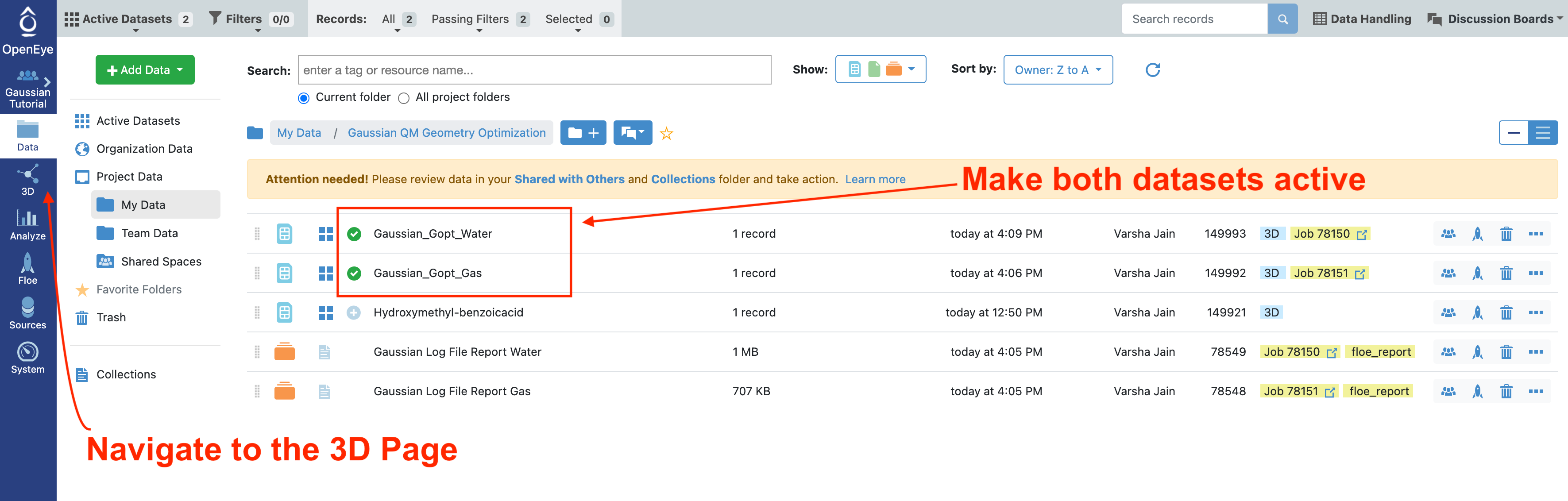
For this tutorial, there should be only one output dataset for each job, Gaussian_Gopt_Gas and Gaussian_Gopt_Water, corresponding to the output for successful calculations. If a conformer or molecule fails the calculation, then a second dataset (Gaussian_Gopt_Gas_Failures or Gaussian_Gopt_Water_Failures) will be created as well. This should not occur for the provided input.
Locate the folder in the data page where the output from both jobs are stored. Select the Plus sign on the Gaussian_Gopt_Gas and Gaussian_Gopt_Water datasets to make them both active.
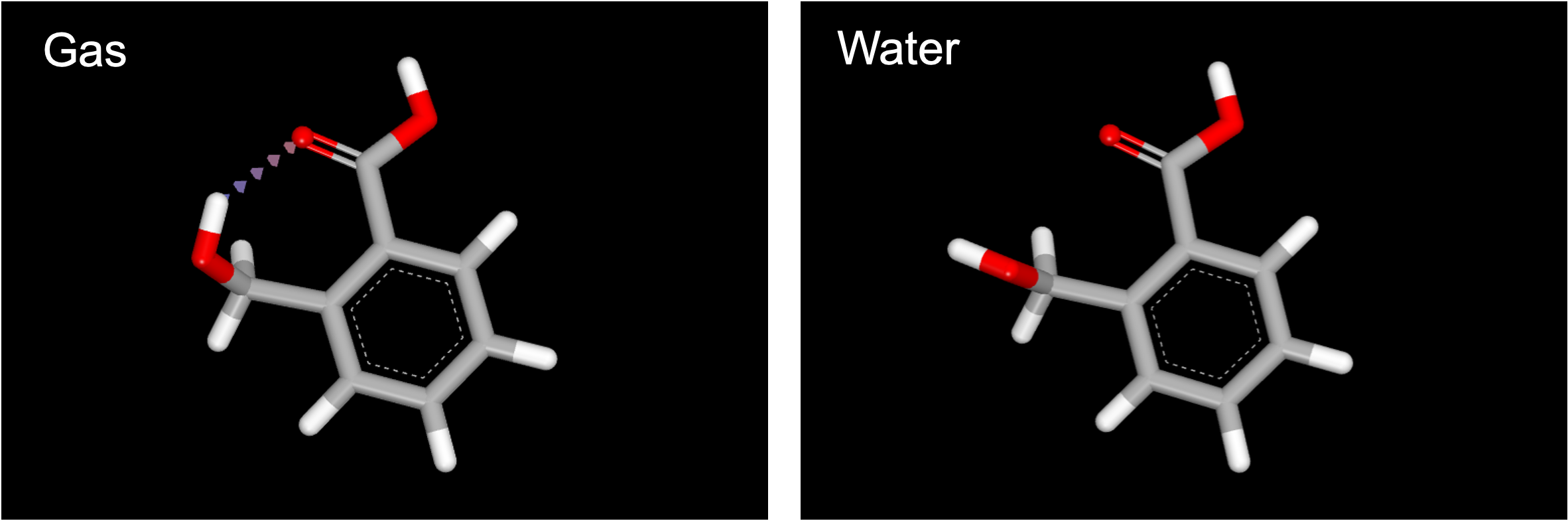
Navigate to the 3D Viewer, where both datasets will be visible on the left side under ALL DATA. Start by selecting the molecule from the Gaussian_Gopt_Gas dataset. When this conformer appears in the viewer, you will see an intramolecular hydrogen bond between the alcohol hydrogen and the carbonyl oxygen. Next, select the molecule in Gaussian_Gopt_Water: in this conformer, there is no internal hydrogen bond. While this is a toy example, there are many benefits to the ability to compare optimized conformer geometries in different environments.