Gaussian QM Single-Point Energy Tutorial
Running this floe for two conformers of benzoic acid will cost around $0.35. Navigate through the categories Product-based / Quantum Mechanics / Gaussian to find this floe. Then, locate or search for Gaussian QM Single Point Energy.
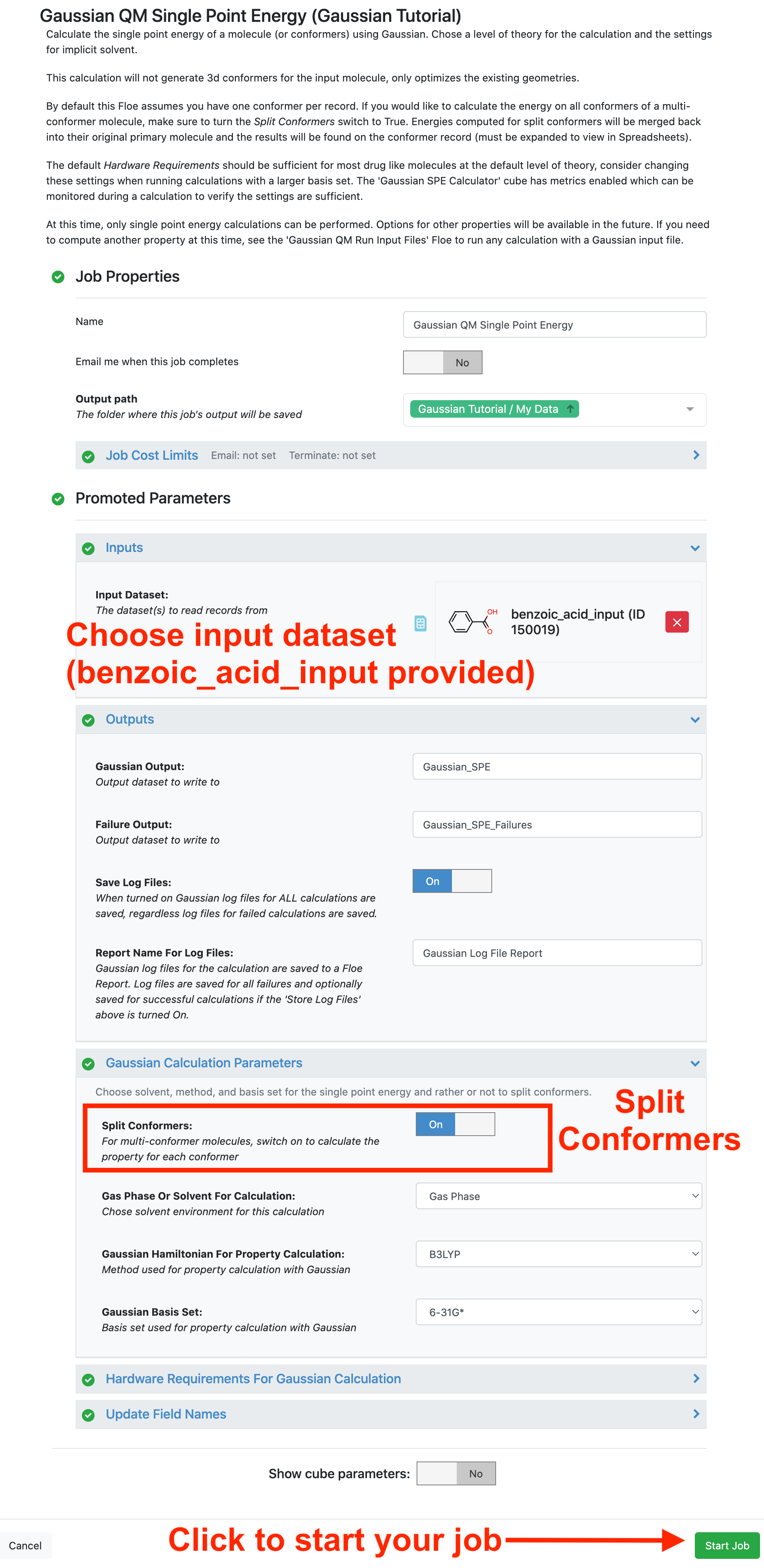
This floe requires 3D input molecule(s) or conformers as input to perform a QM single-point energy calculation using Gaussian. The example input dataset (benzoic_acid_input) is available in the Gaussian QM Single Point Energy folder of the Tutorial Data on Orion. It has two conformers of benzoic acid, the first with the carbonyl group in plane with the aromatic ring and the second with it out of plane.
By default, this floe assumes you have one conformer per record. If you would like to calculate the energy on all conformers of a multiconformer molecule, as in this tutorial, make sure to turn the Split Conformers switch to “On.”
By default, this floe uses B3LYP method and 6-31G* basis set. This combination is chosen to perform calculations with acceptable accuracy at relatively low cost. You can choose the method and basis set, but it will affect the cost. There is also an option for gas phase or implicit solvent. For this tutorial, we will use the default gas phase calculation.
The default Hardware Requirements should be sufficient for most drug-like molecules at the default level of theory (and most small or medium basis sets). The metrics can be monitored on the Gaussian SPE Calculator Cubes to verify the settings are sufficient.
Navigate to the Jobs tab and select this running floe to monitor the status of the calculation.
Note
At this time, only single-point energy calculations can be performed. Options for calculating other properties on datasets will be available in future releases.
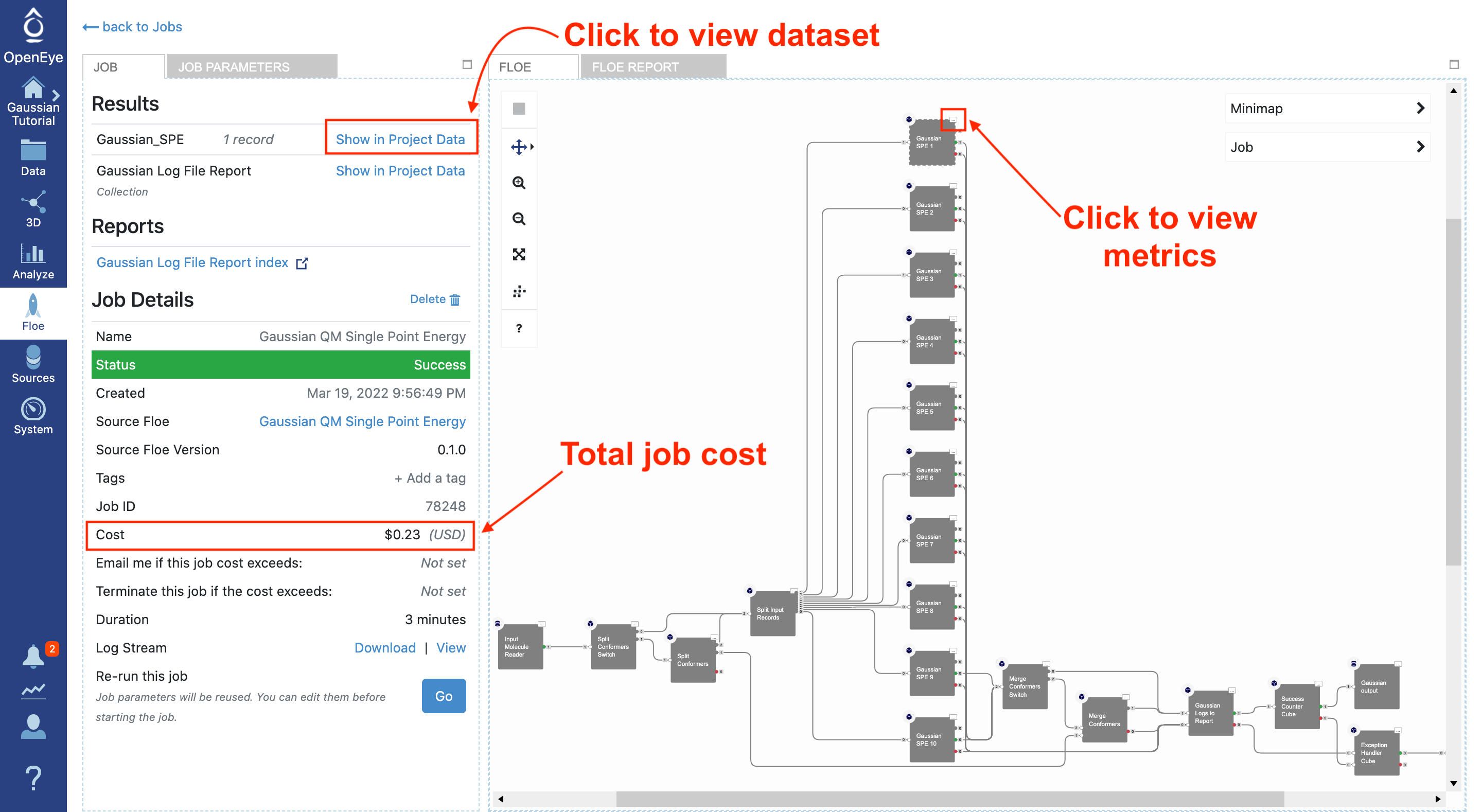
For this tutorial, there should be only one output dataset (Gaussian_SPE) with the single-point energies for successful calculations. If a conformer or molecule fails the calculation, then a second dataset (Gaussian_SPE_Failures) will be created as well – which should not occur for the provided input.
When the floe is complete, the final cost and wall clock time are shown on the left. Listed at the top are the output datasets and reports.
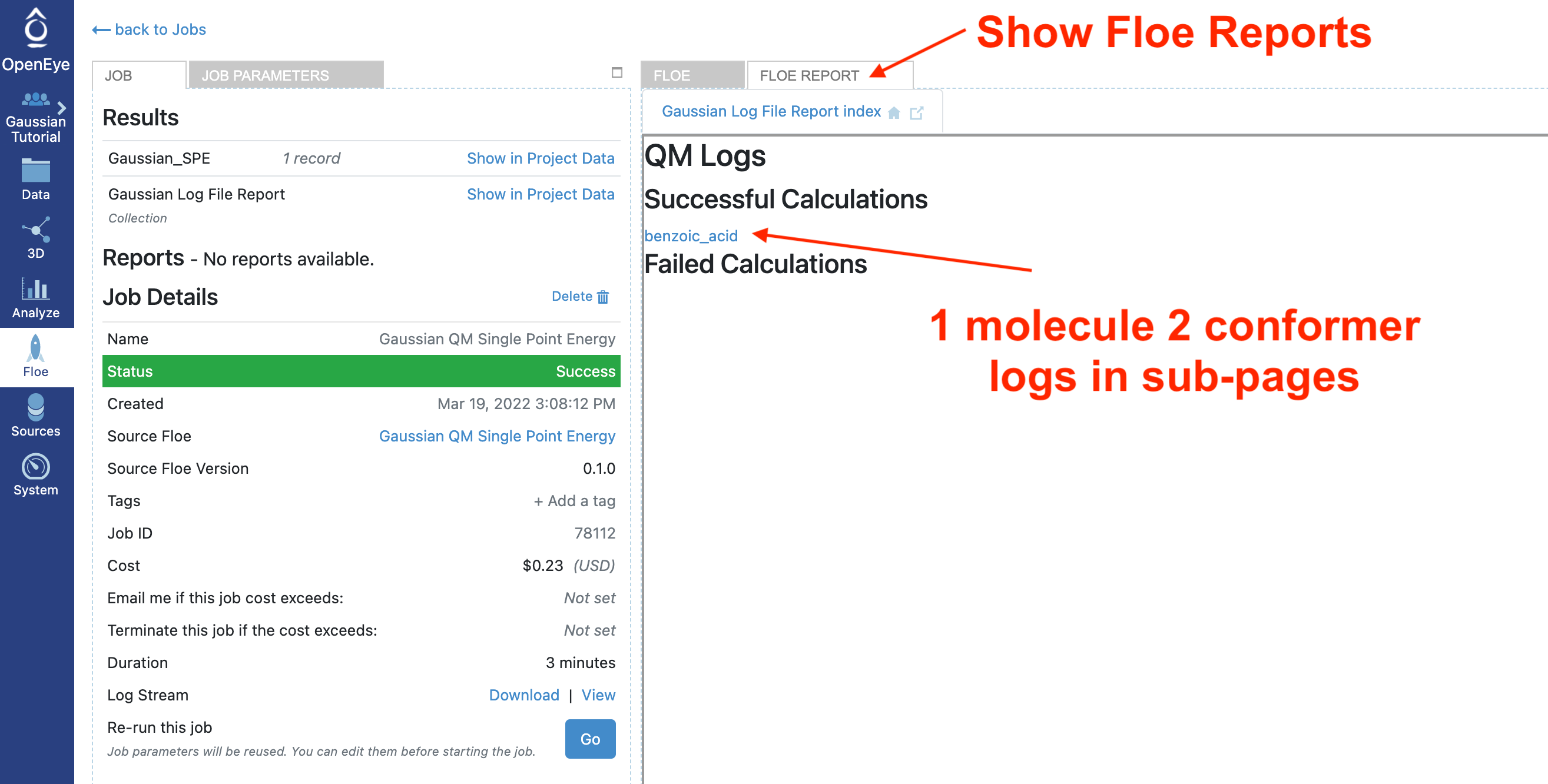
This floe also writes Gaussian log files to a floe report. It separates the logs for successful and failed calculations and links the individual logs to the associated record. This can be useful in understanding unexpected results.
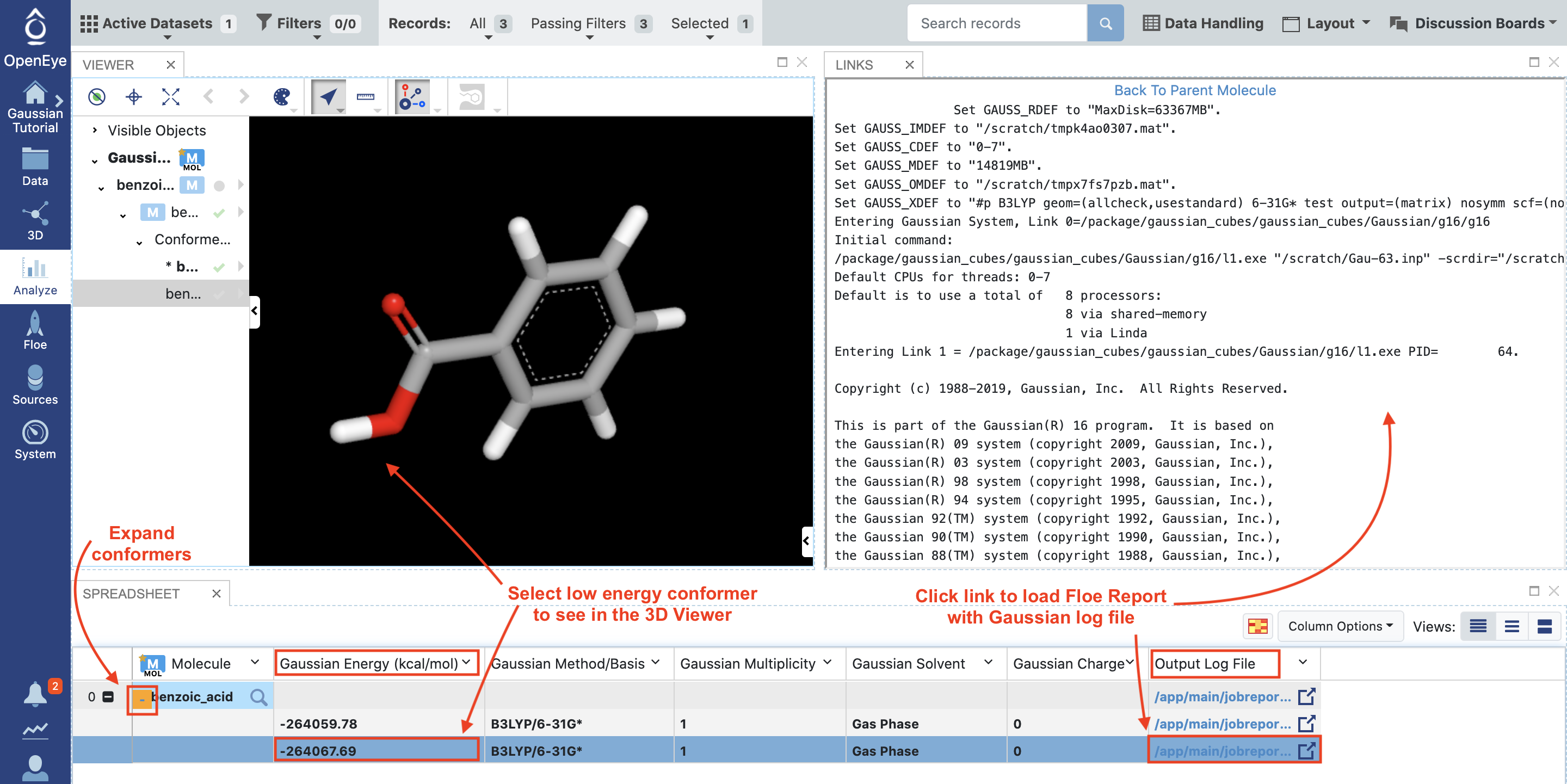
To view the results from this floe, we need to make the Gaussian_SPE dataset active, navigate to the Data page, and select the Plus sign to make this dataset active.
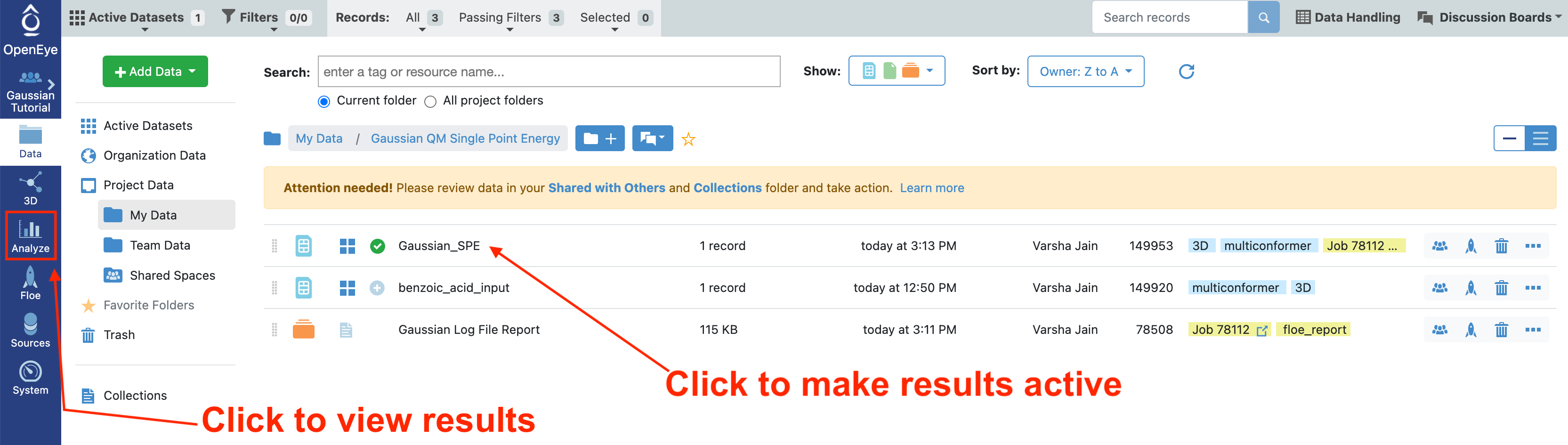
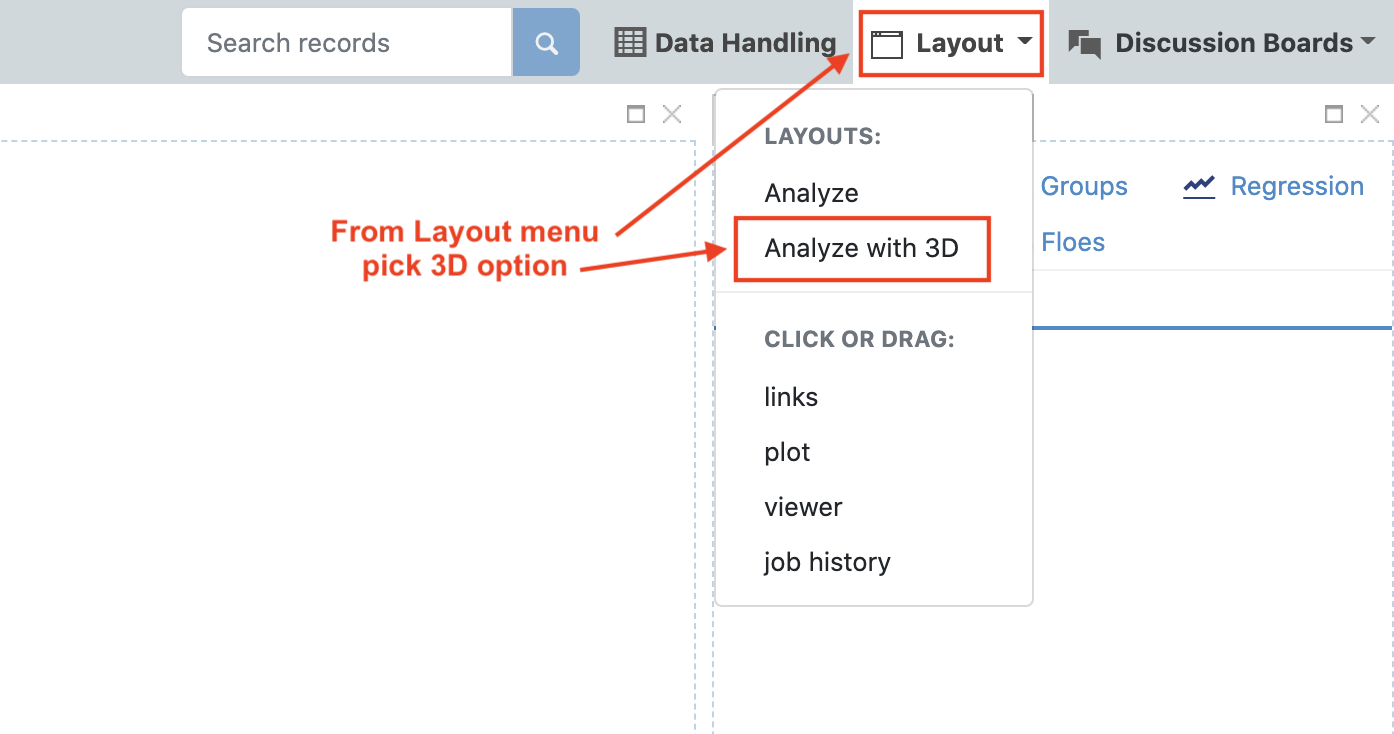
Navigate to the Analyze page and select the “Analyze with 3D” option from the display drop down menu. We do not need the plot for this analysis, so that can be closed.
In the spreadsheet, click the orange Plus sign next to the primary molecule (left column) to expand the conformer records. Click on the lower energy conformer, based on the Gaussian Energy (kcal/mol) column. In the 3D viewer, you can see that the conformer with the carbonyl group in plane with the benzene ring is lower in energy than the conformer with it out of plane. Selecting the link from the Output Log File column will show the Gaussian log file for the calculation on that conformer.