Release Notes, Version 2025.3.1
Small Molecule Discovery Suite Highlights
Structural Biology Floes
The new Structural Biology Floes are used to refine Cryogenic Electron Microscopy (cryo-EM) results to protein conformer ensembles. Cryo-EM captures information about the biomolecules’ native conformational ensemble. The floes in this package leverage weighted ensemble molecular dynamics (WEMD) to explore the full protein conformer space defined by the cryo-EM density maps, or mean maps and eigenmaps when available. Even low-resolution maps guide exploration via global shape, enabling the generation of diverse conformers consistent with experimental evidence.
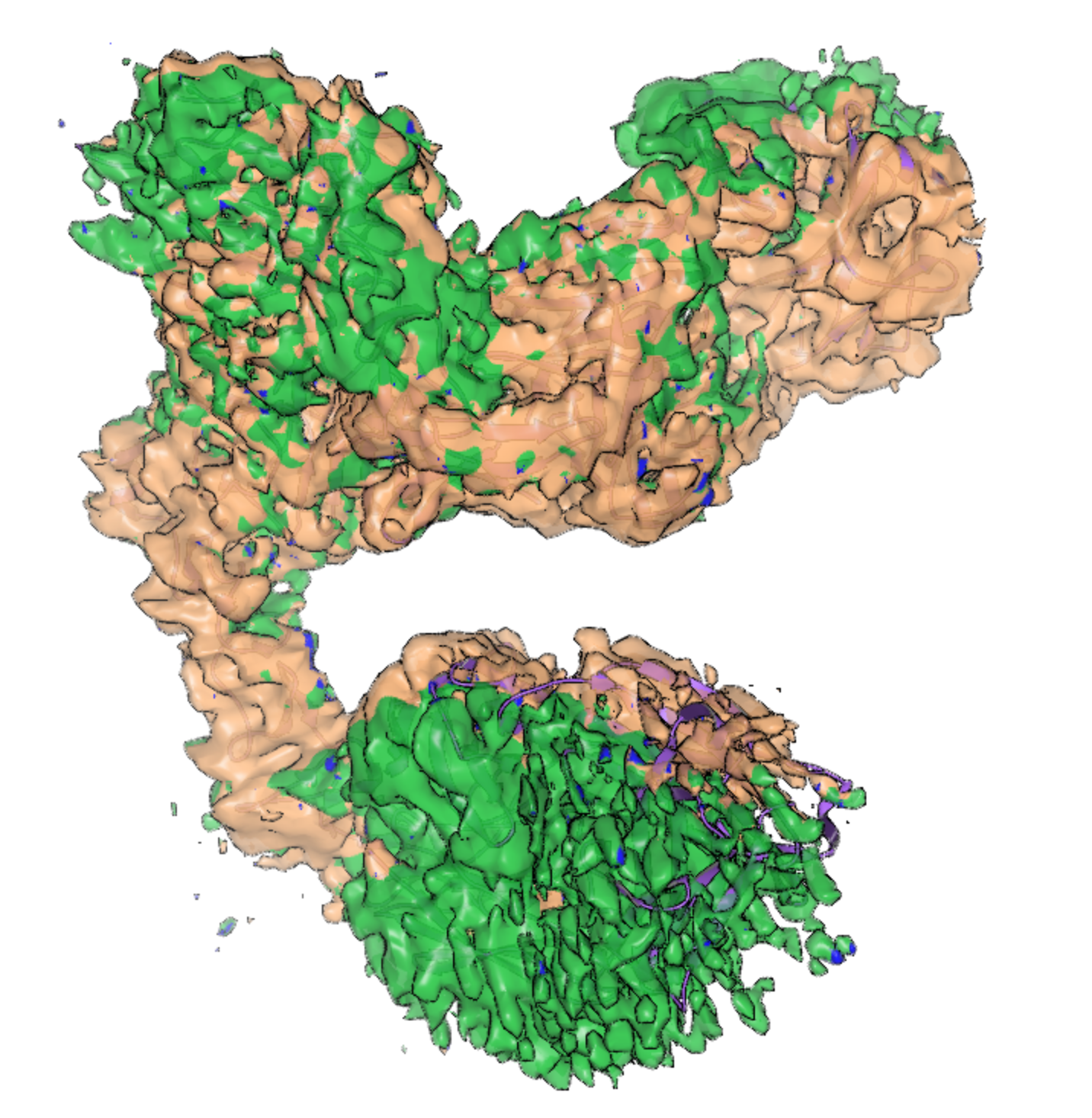
Figure 1. A sample initial protein structure (purple) with cryo-EM density maps, which are used for input in the simulation. The mean map is in blue, and the two heterogeneous maps are in tan and green.
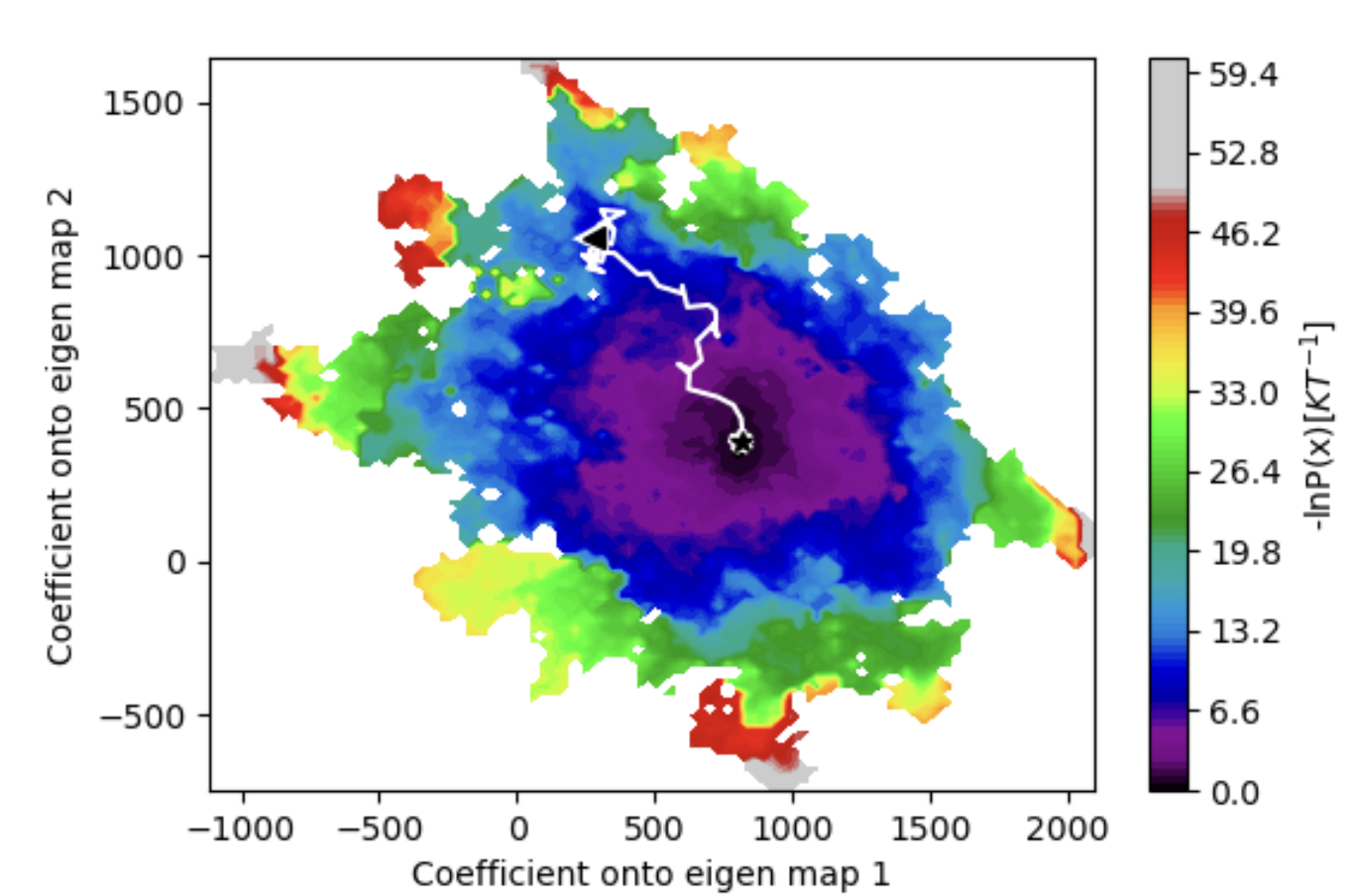
Figure 2. The energy landscape after running weighted ensemble molecular dynamics based on two provided eigenmaps. The metastable state was chosen and an example trajectory is shown for the transition from the starting state to that conformer.
Simulations can be analyzed to identify the best conformers which fit into any provided cryo-EM density maps, even those not used to drive the original WEMD simulation. The resulting energy landscapes can be used to identify metastable states and the transition trajectory between them.
These floes are an affordable way to sample large protein motions, such as secondary structure reorientations or domain motions, potentially requiring only a few WEMD iterations. Each additional density map will be screened against the same simulation to find the best fitting structures; up to dozens of maps can be used simultaneously with minimal added cost.
Output from these floes are all-atom protein conformers which are suitable as input for other calculations, including cryptic pocket detection or SiteHopper.
ML Model Building Floes
ML Build: Graph Convolution Model on Pregenerated Features for Small Molecules
Two new floes are introduced in this release to build graph convolutional neural network (GCNN) models and to use those models for predictions. GCNNs are desirable models primarily because they can learn directly from the molecular graph using atom and bond features from OpenEye toolkits rather than features derived from the molecule (e.g., fingerprints), resulting in fewer parameters for the model to learn. GCNNs are also not limited to interaction radii from complex structures, therefore building more generalizable models.
The new floes are ML Build: Graph Convolution Model on Pregenerated Features for Small Molecules and ML Predict: Graph Convolution Model Prediction. To support these new floes, the ML Build and Predict: Optimal Property-Predicting Graph Convolutional Neural Network Model tutorial and the How to Build and Predict Optimal Property-Predicting Graph Convolutional Neural Network Machine Learning Models by Tweaking Neural Network Architecture how-to-guide have been added to the documentation.
Output analysis in these floes now includes information about how the number of parameters in the model compares to the amount of data. Warnings are included if overfitting based on this ratio is a concern.
Legacy Release Notes
v0.3.2 September 2025
General Notice
This package is built using
OpenEye-toolkits==2025.1.1,OpenEye-orionplatform==6.7.0,OpenEye-Snowball==0.30.0, andOpenEye-orionmdcore==2.5.4.
Minor Changes
Floes now assign missing atomic radii directly to make them more robust to a variety of protein preparation methods.
The Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes have been improved to use the normalized ligandability score in their pocket filtration.
The Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes now correctly output multiple holo design units superposed with the pocket receptors.
The Run a Normal Mode-Guided Weighted Ensemble MD Simulation Floe was previously titled the Run a Weighted Ensemble MD Simulation Floe. Similarly, the Continue a Normal Mode-Guided Weighted Ensemble MD Simulation Floe was previously titled the Continue a Weighted Ensemble MD Simulation Floe. These changes have been made to specify that these floes use protein normal modes as progress coordinates for driving the weighted ensemble simulations.
v0.3.1 August 2025
General Notice
This package is built using
OpenEye-toolkits==2025.1.0,OpenEye-orionplatform==6.4.1,OpenEye-Snowball==0.30.0, andOpenEye-orionmdcore==2.5.4.
Minor Changes
The Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes will finish with the job status “Success” if no cryptic pockets are detected for a given target instead of a “Failed” completion. A Failure Report will be generated listing the potential reasons and next recommendations when no pockets are detected for a target.
If users provide optional Important Residues input while running the Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes, the center-of-mass distance between the user-defined Important Residues and Pocket Residues will be reported for each pocket in the Pocket Receptors output dataset generated by these floes.
Major Changes
The Filter Trajectory Features Data Cube (previously named Preprocess Trajectory Analysis Data) has a seven-to-tenfold lower memory requirement. The Curate Cryptic Pockets Cube (previously named Cryptic Pockets Reporter Cube) has more than a twofold lower memory requirement. Collectively, these changes reduce the chances of failure due to memory error associated with cryptic pocket floe report generation while running the Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes.
The Dynamic Probe Binding Analysis is more than twelvefold faster in this version. The run cost for this floe is now expected to be fivefold less than the previous version.
A new floe, Combined Probe Binding Site Analysis, that combines Probe Occupancy Analysis and Dynamic Probe Binding Analysis has been introduced. Running this floe eliminates the need to separately run the Probe Occupancy Analysis and Dynamic Probe Binding Analysis Floes for cryptic pocket detection.
A new OEligandability-score based pocket ranking has been introduced. For each pocket reported by the Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Combined Probe Binding Site Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes, the total ligandabilty score; the ligandability score normalized by total surface points as well as by receptor volume; and the ligandability significance score will be reported in the Pocket Receptors output dataset.
v0.2.7 March 2025
General Notice
This package is built using
OEToolkits 2024.1.1,Orion Platform 6.1.2, andSnowball 0.28.0,Orion Molecular Dynamics (MD) Core 2.4.2.
Minor Changes
The Cryptic Pockets Reporter Cube has a significantly lower memory requirement. This reduces the chances of failure due to memory error associated with cryptic pocket floe report generation while running the Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes.
Temporary checkpoint files and collection are deleted after the cryptic pocket analysis floes finish. This includes an auto-cleanup for the Automated Cryptic Pocket Detection with Probe Occupancy Analysis, Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes.
v0.2.5 September 2024
General Notice
This package is built using
OEToolkits 2024.1.1,Orion Platform 6.1.2, andSnowball 0.28.0,Orion Molecular Dynamics (MD) Core 2.4.2.
Minor Changes
A fix has been made to allow analysis of protein structures with protonated histidine residue names (HIE, HID and HIP).
An issue with display of pocket residues in the cryptic pocket analysis floe report has been fixed. It now displays the pocket residues when the protein chain IDs do not begin with “A” or when the protein chain IDs do not follow the alphabetical order.
v0.2.4 July 2024
General Notice
This package is built using
OEToolkits 2024.1.1,Orion Platform 6.1.2, andSnowball 0.28.0,Orion Molecular Dynamics (MD) Core 2.4.2.
Minor Changes
A correction has been made in the axis of the 2D free energy projection on the normal modes.
A default value for mutual information cut-off for pocket detection using the Dynamic Probe Binding Analysis and Exposon Analysis Floes is set to 0.00. This parameter is also now exposed to users under Cube Parameters.
Faster receptor creation in the Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis Floes makes them more time efficient.
The documentation now includes an FAQ section.
v0.2.2 April 2024
General Notice
This package is built using OEToolkits 2023.2.3, Orion Platform 6.1.2, and Snowball 0.27.1.
Note
There is a known incompatibility between datasets created by previous versions of Cryptic Pocket Detection and datasets used by this version. If you need assistance with this issue, please contact Support@eyesopen.com.
Major Changes
The Automated Cryptic Pocket Detection with Probe Occupancy Analysis Floe is introduced for automated, end-to-end workflows for cryptic pocket detection with probe occupancy analysis using an input design unit prepared using Spruce.
Three advanced Floes (Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis) automate the cryptic pocket analysis process by integrating analysis of weighted ensemble protein sampling data, iterative clustering, cryptic pocket detection, and receptor creation.
Weighted ensemble protein sampling data storage has been significantly refactored, which reduces the overall size by half and supports faster data find and read.
Two new options have been added, Maximum Number of Selected Modes and Mode Filtering Property, in the Calculate Normal Modes Floe that allow for automatic rank and select modes based on specific mode properties. This feature is turned On by default in the Automated Cryptic Pocket Detection with Probe Occupancy Analysis Floe.
Minor Changes
In addition to the Automated Cryptic Pocket Detection with Probe Occupancy Analysis Floe, individual sub-floes starting from the Solvate and Equilibrate Target Protein Floe to three cryptic pocket analysis Floes (Probe Occupancy Analysis, Dynamic Probe Binding Analysis, and Exposon Analysis) can be sequentially run for protein sampling and cryptic pocket analysis.
Now the Run a Normal Mode-Guided Weighted Ensemble MD Simulation Floe generates and stores data in a V2 ShardCollection.
The outputs have been changed from the Run a Normal Mode-Guided Weighted Ensemble MD Simulation Floe. Now the output dataset contains multiple records (one record per iteration) and only contains summary statistics for the simulation. All simulation data (e.g., weighted ensemble data, MD trajectories, simulation files) are stored in the output shard collection. The collection is now self-contained and required as input for downstream analysis Floes.
The Subsystem Selection String option has been substituted with Environment Selection String in the Calculate Normal Modes Floe for ease of use. Now the system-environment framework is enabled by default in the Floe.
The Continue a Normal Mode-Guided Weighted Ensemble MD Simulation Floe now takes the collection generated by the Run a Normal Mode-Guided Weighted Ensemble MD Simulation Floe as the input to resume the weighted ensemble protein sampling simulation.
v0.1.2 September 2023
General Notice
This package is built using OEToolkits 2023.1.0, Orion Platform 5.1.1, and Snowball 0.26.0.
Minor Changes
Fixes an issue related to “select string” parameter format in the Calculate Normal Modes floe.
Enables support for non-canonical amino acids in the Solvate and Equilibrate Target Protein floe.
A minor typographical correction in documentation for the Calculate Normal Modes floe.
v0.1.0 July 2023
General Notice
This is the first release of OpenEye Cryptic Pocket Detection Floes.
This package is built using OEToolkits 2023.1.0, Orion Platform 5.1.0, and Snowball 0.26.0.
Legacy Release Notes
v6.6.0 August 2025
General Notice
This package was built using
OpenEye-orionplatform==6.6.0andOpenEye-toolkits==2024.1.1.
Functional Changes
The records_per_shard parameter on the ParallelRecordsToCollectionCube cube has been promoted in the Dataset to Collection Export ETL floe.
v6.3.2 January 2025
V6.3.2 is a rerelease of 6.3.1, with no change in content.
v6.3.1 September 2024
A bug has been addressed in which SMILES data in an input file following CXSMILES data caused a parsing error.
A bug has been addressed when exporting datasets to CSV files. All columns, or a subset thereof, can now be exported in cases when previously only Molecule and Molecule.Titled would have been exported.
v6.3.0 August 2024
A bug has been addressed in which SMILES data in an input file following CXSMILES data caused a parsing error.
v6.2.0 June 2024
Support for ShapeQuery (.sq) files has been added.
Support for CXSMILES data has been added.
Created collections have been defaulted to v1.
A bug has been addressed by which some english language words (such as “BOTH”) could be interpreted as molecules.
v6.1.2 February 2024
Columns can now be optionally ignored when exporting datasets.
v6.0.0 September 2023
The version number has been synced with openeye-orionplatform.
The Dataset to File Export ETL floes now preserve the Molecule Title when exporting to CSV files.
v2.3.0 July 2023
This version increases the minimum versions of the requirements:
OpenEye-orionplatform==5.1.0openeye-toolkits>=2022.2.2
v2.1.3 February 2023
A floe for uploading large or small files from a URL has been added
v2.1.2 November 2022
The orion-platform requirement has been updated to 4.5.2
v2.1.1 September 2022
The orion-platform requirement has been updated to 4.4.1
An issue where
orionclient.types.Shard.download()could receive less data than expected has been fixed.An
IOErrorwill be raised andorionclient.types.Shard.download_to_file()will catch and retry.
v2.1.0 July 2022
An issue with importing convert_path has been fixed.
The etl-floe classifications has been updated to use the new
PurePosixPathclassificationsThe
brieffield has been added and thedescriptionfield has been made more descriptive on all ETL floes.Requirement changes:
The Sphinx requirement has been updated to
3.5.4and moved to requirements.txt.The jinja2 requirement has been updated to
3.0.3.The orion-platform requirement has been updated to
4.4.0.The openeye-toolkits requirement has been updated to
2022.1.1.The openeye-floe-pkg-tools requirement minimum version has been updated to
1.0.3.
The etl-floe classifications has been updated to use the new
PurePosixPathclassifications.The “brief” field was added and the “description” field was made more descriptive on all ETL floes.
v2.0.2 February 2022
Updated orion-platform to 4.3.0.
Converting datasets containing image data to csv now results in that field containing “<Image>”.
v2.0.1 December 2021
The openeye-toolkits requirement has been updated to 2021.2.0
v2.0.0 November 2021
The Collection Resize floe has been removed
v1.2.9 November 2021
The orion-platform requirement has been updated to 4.2.5
v1.2.8 October 2021
The orion-platform requirement has been updated to 4.2.4
A bug where the metadata of a collection was not being preserved when running the Collection Resize ETL floe has been fixed.
A required parameter collection_type in the ParallelCollectionResizeCube of the Collection Resize ETL floe has been promoted.
v1.2.7 October 2021
orion-platform has been updated to 4.2.0
A failure caused by csv files containing no data for some columns has been fixed.
IUPAC names are no longer parsed, resulting in fewer column splits and ETL speedup.
A failure parsing lists of numbers has been fixed.
A bug in the naming of split columns has been fixed.
A failure when converting a record to OEB has been fixed. Fields with data that cannot be properly attached as generic data to a molecule are now skipped by default.
v1.2.6 June 2021
The orion-platform requirement has been updated to 4.1.0 (mono-package)
The OpenEye-toolkits requirement has been updated to 2021.1.1
A regression in perceiving basic molecular properties for SMILES and CSV has been fixed.
v1.2.5 June 2021
The orion-platform requirement has been updated to 4.0.0 (mono-package)
Orion Platform >= 4.0.0 will not work with stacks older than 2021.1 due to usage of new APIs
The OpenEye-toolkits requirement has been updated to 2020.2.4
A new Converter,
SmilesFileConverter, has been added.When converting CSV and SMILES format files to records, the original smiles are stored on output records.
ETL floe documentation has been improved
v1.2.4 November 2020
The orion-platform requirement has been updated to 3.1.0
BaseDatasetFieldAddOrReplaceCubehas been added.If no file extension is provided to
DatasetWriterCube’s output file parameter, the file format is nowOEDB.OpenEye-floe==0.12.0
Adds support for versioning of WorkFloes.
Adds support for ordering of Parameters in the Orion UI.
An issue that resulted in Serial Cube Group initializer ports not incrementing the counts has been resolved.
OpenEye-drconvert==1.1.2
Bump OpenEye Toolkits
OpenEye-orionclient==3.1.0
Bump OpenEye Toolkits
OpenEye-datarecord==3.0.1
Bump OpenEye Toolkits
v1.2.3 August 2020
orion-platform has been updated to 3.0.0
An issue with too frequent flushing in
DatasetUpdaterCubehas been fixed.The
DatasetAppenderCubenow finalizes datasets.OpenEye-datarecord==3.0.0
The implementation of datarecord has mostly moved to C++ in the toolkits.
A field type
DesignUnitfor storing OEDesignUnits has been added.
OpenEye-drconvert==1.1.0
Support for OEDesignUnit has been added
OpenEye-orionclient==3.0.0
Added OEDesignUnit support for dataset upload and download
Support for uploading OEDU files to datarecords has been added to
upload()orionclient.types.Datarecord.recordsnow yieldsOEMolRecord
v1.2.2 April 2020
orion-platform has been updated to 2.4.5
OpenEye-orionclient==2.6.3
The efficiency and reliability of
ShardCollection.list_shardshas been improved.
v1.2.1 March 2020
orion-platform has been updated to 2.4.4
A bug in
RecordsToCollectionCubehas been fixed.Improvement made to shard retries.
v1.2.0 February 2020
The base OS image has been upgraded to Amazon Linux 2
The environment has been upgraded to Python 3.8.1
Package documentation has been added
orion-platform has been updated to 2.4.2
Improvements since 2.1.0 include:
Support for the
OEZformat has been added to the following cubes:CollectionResizeCubeCollectionToRecordsCubeRecordsToCollectionCube
orionplatform.cubes.files.RecordsToRecordFileConverternow uploads its files in parts reducing the disk usageNew parameters, which optionally enable metric collection for cubes have been added.
Sets lower bound on OpenEye-toolkits>=2019.10.2
A duplicate of
output_tagsin theDatasetWriterCubeOptions parameter group has been removed.OpenEye-Drconvert==0.7.2
Support added for Float and Int vectors
Drconvert CLI now supports going from records to mols
v1.1.1 October 2019
orion-platform 2.1.0
Improves reliability of uploading shards and writing shards of records
Adds
limitparameter toShardReaderCubeto allow streaming only some shards from a collection.
v1.1.0 August 2019
orion-platform 2.0.0
Improves reliability of collection floes
Updated version of drconvert (0.6.20) correctly handles conformer data
v1.0.0 July 2019
orion-platform 1.0.2
v0.1.29 April 2019
orion-platform 0.1.16
Collection resize floe
v0.1.28 February 2019
Bugfixes
drconvert 0.6.16
No longer sets
OEIsomericConfTeston file formats that are not mol2 or sdf
v0.1.27 February 2019
Add Dataset Copy floe
v0.1.26 February 2019
Orion Platform 0.1.14
openeye-drconvert>=0.6.15,<0.70
Performance improvements
Bugfixes
Fixes
ArchiveConverterCube
v0.1.25 January 2019
Orion Platform 0.1.13
openeye-drconvert>=0.6.13,<0.70
Calls
flush()before returning records fromMolFileConverter
openeye-datarecord>=0.12.7,<0.13.0
Adds
flush()toOERecordto write all cached values to bytes.
openeye-schemagen>=0.3.3,<0.4.0
Minor cleanup
v0.1.24 December 2018
orion-platform 0.1.12
Summary of changes relevant to ETL floes:
openeye-drconvert>=0.6.12,<0.7.0
Handle whitespace in SD data
openeye-orionclient>=0.6.10,<0.7.0
Makes Orion Client more robust to handling API changes
v0.1.23 November 2018
orion-platform 0.1.11
v0.1.22 November 2018
orion-platform 0.1.10
Summary of changes relevant to ETL floes:
openeye-schemagen>=0.3.2,<0.4.0
survive SD data consisting of only whitespace
openeye-datarecord>=0.12.5,<0.13.0
add more testing for metadata; Serialize/deserialize round trips. ORION-6328
add Meta.Flags.Predicted metadata to datarecord. Minor cleanups. ORION-6328
v0.1.21 October 2018
orion-platform 0.1.9
Summary of changes relevant to ETL floes:
openeye-schemagen>=0.3.1,<0.4.0
delimiters back to ordering by descending priority
openeye-orionclient>=0.6.9,<0.7.0
0.6.9
Adds headers to improve debugging capabilities
0.6.8
Adds orionclient.exceptions.OrionTimeout exception that indicates that
a request couldn’t be made to the specified Orion within the expected time
openeye-floe>=0.6.15,<0.7.0
0.6.15
Fixes issue where parallel workers would incorrectly propagate failures when run in Orion
v0.1.20 October 2018
add package uuid
v0.1.19 October 2018
orion-platform 0.1.8 * fix memory leaks when reading dataset * improve robustness of collection handling
v0.1.18 September 2018
orion-platform 0.1.7 orion-client fixes: * avoid writing too many records at once * fix issue retrieving records when sizes of records are not consistent
v0.1.17 September 2018
orion-platform 0.1.6
v0.1.16 September 2018
sync job system tags with backend/frontend
bump orion-platform to 0.1.5 to force min versions
remove default tag that appears to not be used
Added continuation char BUGZID: ORION-6089
Allowing non-SDData formats for etl export BUGZID: ORION-6089
v0.1.15 September 2018
Needed to rev version because of Orion failing inspection due conflict with a previous failed upload of this version:
“Workfloe Record Collection to Dataset with version:’0.1.14’ and uuid:’934d77a2-c1a1-421f-8daf-5b8ca6325fbf’ already exists for user with ID 1”
v0.1.14 September 2018
Add package setup, tasks, tests, and release notes
invoke upload-packageputs package in S3 for devops to load onto stacksUpdate orion-platform requirement to 0.1.4, which pulls in toolkit, drconvert and related packages
Add record collection floes
Legacy Release Notes
This package is built using OpenEye-orionplatform==6.2.0, OpenEye-toolkits==2024.2.0, OpenEye-Snowball==0.28.1, and OpenEye-floereport==6.2.0.
There is now a baseline XGBoost model with every ML Build floe. The performance of these XGBoost models is reported in the Floe Report and output success model.
Several bug fixes have been made.
ML Predict: XGBoost for Small Molecules Floe
The XGBoost models built by default using ML Build floes can be used as input here. This floe can be used to predict the properties of small molecules using XGBoost from the ML Build floes. It will act as a classifier if the base model is a classifier trained on String and will act as a regressor if trained on Float. The floe autoselects the type of prediction based on the provided model. If the model was trained on a custom feature vector, please provide a field in the input molecule dataset containing the feature vector; otherwise, the floe will fail.
This package is built using
OpenEye-orionplatform==6.2.0,OpenEye-toolkits==2024.1.1,OpenEye-Snowball==0.28.0, andOpenEye-floereport==6.2.0.All Floes have new tutorial links.
Scaffold splitting has been added to the Data Processing of Small Molecules for ML Model Building Floe.
The option to use a Keras Hyperparameter Tuner has been added to the ML Build: Regression Model with Tuner using Feature Input Floe.
A default Adjust Batch Size parameter has been added to the Advanced: Neural Network Hyperparameter Options for all ML Build Floes.
This release includes a new style for Floe Reports, bug fixes, and more stable Floe runs.
This package is built using
OpenEye-orionplatform==5.1.0,OpenEye-toolkits==2023.1.0, andOpenEye-Snowball==0.26.0.All Floes have new brief texts and Floe categories.
Keras Hyperparameter Tuner.
This is an optimizer for fingerprint based model building (classification and regression).
Choose from Hyperband, RandomSearch, and Bayesian Optimization.
It is available in Floes: ML Build: Regression Model with Tuner using Fingerprints for Small Molecules and ML Build: Classification Model with Tuner using Fingerprints for Small Molecules.
This release includes a new style for Floe reports, bug fixes, and more stable Floe runs for larger data.
hERG Toxicity Prediction for Small Molecules using ML and Cheminfo Fingerprints.
This Floe predicts and explains hERG toxicity of molecules as either “High” or “Low.”
ML ReBuild: Transfer Learn ML Regression Model using Fingerprints for Small Molecules.
This Floe is for Transfer Learning on previously built Machine Learning Models in Orion.
This is the first minor release of Model Building package with package version 0.10.4.
This package is built using
OpenEye-orionplatform==4.4.0,OpenEye-toolkits==2022.2.1, andOpenEye-Snowball==0.24.0..Added Overfit correlation and coefficients [refer to Optimization Strategy V] to train models without overfit.
Added Precision, Recall, Specificity, and Sensitivity parameters in floe report for Classification Model Building Floes. These fields are also available in record output.
This is the second major release of Model Building package with package version 0.10.0.
This package is built using
OpenEye-orionplatform==4.5.4,OpenEye-toolkits==2022.2.1, andOpenEye-Snowball==0.24.0..All floes have a new brief parameter and floe categories.
ML Build: Classification Model with Tuner using Fingerprints for Small Molecules trains multiple Neural Network Classification models on physical properties of small molecules. Uses cheminformatics and machine learning to train said models.
ML Predict: Classification using Fingerprints for Small Molecules predicts discreet classes from a pretrained neural network model. Needs molecule database and neural network model dataset as input (trained using the previous floe).
ML Build: Regression Model with Tuner using Feature Input trains multiple Neural Network Regression models on physical properties of small molecules. Uses user provided custom feature vector and machine learning to train said models.
ML Predict: Regression using Feature Input predicts property from a pretrained neural network model. Needs molecule database, feature vector(same length as training data), and neural network model dataset as input (trained using the previous floe).
Data Processing of Small Molecule for ML Model Building analyzes and preprocesses data for training ML(machine learning) models. Needs molecule database and response value (either for classification or regression).
ML Regression Model Building using Fingerprints for Small Molecules (previously Neural Network Based Regression Model Building) trains multiple Neural Network (Full or Probabilistic) Regression models on physical properties of small molecules. Uses cheminformatics and machine learning to train said models.
ML Regression using Fingerprints for Small Molecules (previously Physical Property Prediction for Small Molecules using Machine Learning) predicts property from a pretrained neural network model. Needs molecule database and neural network model dataset as input (trained using the previous floe).
Solubility Prediction for Small Molecules using Machine Learning and Cheminfo Fingerprints predicts solubility in log uM using neural network based machine learning. Needs molecule database as input.
This package is built using
OpenEye-orionplatform==4.4.0,OpenEye-toolkits==2022.1.1, andOpenEye-Snowball==0.24.0..Bug fixed on Physical Property Prediction for Small Molecules using Machine Learning
Model and Record ID in Neural Network Based Regression Model Building renamed for user convenience.
Better use case handling for wrong inputs.
This is the first release of Model Building package with package version 0.9.0.
This package is built using
OpenEye-orionplatform==4.4.0,OpenEye-toolkits==2022.1.1, andOpenEye-Snowball==0.24.0..All floes have a new brief parameter and floe categories.
Neural Network Based Regression Model Building trains multiple Neural Network (Full or Probabilistic) Regression models on physical properties of small molecules. Uses cheminformatics and machine learning to train said models.
Physical Property Prediction for Small Molecules using Machine Learning predicts property from a pretrained neural network model. Needs molecule database and neural network model dataset as input (trained using the previous floe).
Solubility Prediction for Small Molecules using Machine Learning and Cheminfo Fingerprints predicts solubility in log uM using neural network based machine learning. Needs molecule database as input.
Legacy Release Notes
v 6.5.7 September 2025
General Notice
This version pins
orionmdcore==2.5.3.1, where temporary resources are deleted immediately after use.The NES chimera construction has been improved to avoid cube failures during ring fusion involving a cyclopropyl group.
v 6.5.6 March 2025
General Notice
Structured water analysis in the Short Trajectory MD with Analysis and Analyze Protein-Ligand MD Floes is now turned on by default (Process Water On) in the Trajectory To OEMols Cube. If there is no structured water found near the binding site, it will skip the water analysis.
A collection handling issue introduced in the Ligand Bound and Unbound Equilibration for NES Floe in the version 6.5.3 has been fixed.
This version pins
orionmdcore==2.5.3, which includes minor bug fixes, including an implementation of a less memory-intense protocol in several cubes, which fixes the risk of out of memory errors witnessed in version 6.5.3 of MD floes. Detailed release notes of version 2.5.3 can be found here: Molecular Dynamics Core Package Release Notes.The Compare Experimental Affinity with NES Results Floe was removed because it was redundant with other analysis floes.
v 6.5.3 February 2025
General Notice
We now support the interactive floe report for the Edge Mapper for RBFE calculations Floe, which allows a user to modify the perturbation map and save the changes as a dataset on Orion.
A new clustering-based edge map type, a multi-star map, has been added to the Edge Mapper for RBFE calculations Floe, and the star map hub selection scheme has been updated to select the representative parent molecule as a hub.
This version pins
orionmdcore==2.5.1.We now print clear error messages in the Floe Failure Report when a rectangular solvation box is chosen without any restraints (along with a Checker Bypass switch of the Bound Protein Ligand MD Health Checker Cube turned OFF). The floe is stopped with a Failed status when that happens. A distance threshold can also be provided to terminate MD runs of ligands if the ligands move beyond that threshold within the binding site after equilibration. This can be done via the shell_cutoff parameter of the Bound Protein Ligand MD Health Checker Cube. The names of those ligands that experience such egress will be noted in the Floe Failure Report. This feature is available in Equilibration and Nonequilibrium Switching, Plain Molecular Dynamics Floe, Ligand Bound and Unbound Equilibration for NES, Bound Protein-Ligand MD, Short Trajectory MD with Analysis, and Solvate and Run MD Floes.
We allow only one protein molecule as input (not to be confused with multiple chains) to the Short Trajectory MD with Analysis, Ligand Bound and Unbound Equilibration for NES, Equilibration and Nonequilibrium Switching, Solvate and Run MD, Bound Protein-Ligand MD, and Induced-Fit Posing (Confined) Floes. The floes fail quickly when that is found not to be the case.
Floe Updates
The Short Trajectory MD with Analysis and Analyze Protein-Ligand MD Floes now write the ligand binding interaction occupancy CSV file to the analysis result archive file.
The Edge Mapper for RBFE calculations Floe by default sets the number of CPUs to run the OEMapper Cube to 48, in order to speed up the perturbation map generation for the large ligand sets.
The Short Trajectory MD with Analysis Floe carries FreeForm evaluations only when using the FreeForm conformers as unbound sampling starting conformers. By default, the input bioactive conformation is used as a starting conformer for the unbound sampling.
In the Equilibration and Nonequilibrium Switching Floe, independent choices can be made for the solvation box shapes of the bound and unbound states of ligands. Whereas choosing a rectangular box for the bound state without imposing restraints (unless the Checker Bypass is ON) is still prohibited, box shape choice for the unbound state is not restricted by such restraints. This replicates what has been available in the Short Trajectory MD with Analysis Floe.
Users can now tune the frequency at which lines reporting the progress of MD production, equilibration, and warm-up stages are printed. They each have a default value (in ns). These defaults are larger than the previously fixed values. The change produces smaller log files that are easier to browse. This affects the Equilibration and Nonequilibrium Switching, Plain Molecular Dynamics Floe, Ligand Bound and Unbound Equilibration for NES, Bound Protein-Ligand MD, Short Trajectory MD with Analysis, and Solvate and Run MD Floes.
Promoted parameters of the Equilibration and Nonequilibrium Switching, Nonequilibrium Switching Recovery, NES Result Utilities, Nonequilibrium Switching, and Ligand Bound and Unbound Equilibration for NES Floes have been placed in appropriate parameter groups that can be seen in their respective job forms.
Structured water analysis in the Short Trajectory MD with Analysis and Analyze Protein-Ligand MD Floes is turned off by default (Process Water Off) in the Trajectory To OEMols Cube.
Cube Updates
The default setting of holonomic constraints in NES has been changed in the NESGMX Cube. This setting now defaults to Bonds2H (default was previously All-Bonds). Along with some other changes to MD settings in NES, this helps to make the MD more numerically stable in NES switches. Although All-Bonds remains a selectable option, it is not recommended because of numerical instabilities in switches involving large mass changes (e.g., hydrogen to bromine).
A fatal port has been added to the Bound Protein Ligand MD Health Checker Cube for proper exception handling related to box shape choices that are inconsistent with restraints on the solute. Records transmitted through the failure report have also been refined to indicate which ligand moved out of the binding pocket after equilibration.
A Skip the edge scoring option has been added to the OEMapper Cube to enable the fast star map generation when a user specifies the hub ligand(s) of the star map.
v 6.4.4 October 2024
General Notice
Bug Fixing. A memory leak has been found and fixed in the NESAnalysis cube. The bug could hit the NES floes if a large number of edges needed to be processed.
Cube Updates
Fix a memory leak bug in the NESAnalysis cube.
v 6.4.3 July 2024
General Notice
Structured water analysis is now embedded within the Short Trajectory MD with Analysis and Analyze Protein-Ligand MD Floes. This enhancement will analyze explicit solvent MD simulations and identify structured water.
Protein tumbling restraints are now automatically applied during the bound state free energy calculations in the Nonequilibrium Switching Floe if they were requested and successfully applied in the Short Trajectory MD with Analysis Floe and the Ligand Bound and Unbound Equilibration for NES Floe. It also means that tumbling restraints are used throughout the Equilibration and Nonequilibrium Switching Floe, if they are requested and applied successfully. Previously, they were only applied for performing equilibrium simulations and not during free energy calculations.
If protein tumbling restraints are used in NES, pressure is regulated by the Berendsen barostat. If not, then the Parrinello-Rahman barostat is used.
This version pins
orionmdcore==2.3.2, which includes minor Cube parameter definition updates and OpenFF 2.2.0 support.The MD Floes now support OpenFF 2.2.0; it is used as a default force field to be applied to ligands.
Floe Updates
A typo was fixed in a parameter name for the Short Trajectory MD with Analysis Floe. Previously, users submitting an Orion job via the python API would have set the Freeform Output Dataset name using the parameter freefrom_out; this has been corrected to freeform_out.
HTML links were added from floe descriptions to tutorial content where applicable.
The structured water analysis tool has been added to the Short Trajectory MD with Analysis Floe. This analysis will now be performed automatically.
The Analyze Protein-Ligand MD Floe has been updated to include structured water analysis as well.
The Nonequilibrium Switching and Equilibration and Nonequilibrium Switching Floes will now apply protein tumbling restraints if they were applied in an upstream Floe or, in the case of the latter, requested in the input. Pressure will be regulated using the Berendsen barostat if tumbling restraints are in place.
The Nonequilibrium Switching Floe can now accept a Bound Input Dataset generated by the Short Trajectory MD with Analysis Floe, even if the latter ran multiple replicates of simulations per ligand. This is a fix to the bug that allowed inputs with only one replicate per ligand.
The Equilibration and Nonequilibrium Switching Floe performs an early check for the syntax of the input Ligand Experimental Affinity File and shuts the Floe down if bad syntax is detected.
The hub autoselection scheme for the star map has been updated in the Edge Mapper for RBFE calculations Floe. While it is currently under active development to find a good heuristic for the star map hub selection, the ligand with the minimum heavy atom count (HAC) will be selected as a hub ligand for the star map.
The Analyze Protein-Ligand MD and Short Trajectory MD with Analysis Floes now set the trajectory stride for the trajectory analysis based on the size of the flask and the total number of frames to avoid the out-of-memory issue during the analysis.
New Cubes
The Initialize Water Calculation Cube creates a grid of voxels for calculating water and assigns water molecules to nearby voxels.
The Calculate Grid-Based Water Cube calculates water sites based on voxels and maps water molecules from each frame of the simulation to the water sites.
The Analyze Water Cube calculates interactions between water and a ligand, as well as between water and a protein.
The Analyze Clustered Water Cube calculates water interactions based on clustered trajectories.
The Bound Dataset Health Checker Cube screens the Bound Input Dataset in the Nonequilibrium Switching Floe for mixed tumbling restraint weights across simulations of different ligands. If mixing is found, the Floe is failed.
The Affinity File Format Checker Cube checks if the syntax of the Ligand Experimental Affinity File is correct. If found to be incorrect, the Floe is failed.
The Trajectory Stride Setting Cube sets the trajectory stride for the downstream trajectory analysis so that the number of frames times the flask size per ligand does not exceed 200 million.
Cube Updates
A typo has been fixed in the header of the NES output file nes_output_dataset_result.tar.gz created by the NESResultsToFile Cube. Previously, the header listed DD4G; this has been corrected to DDG. This change should only affect scripted parsing done outside Orion in a manner that relies on the DD4G tag.
A logical error has been fixed in the NESGMXChimera Cube that previously led NES to ignore user-set number of switches and to always run 80 switches when the input datasets came from the Ligand Bound and Unbound Equilibration for NES Floe. This issue did not previously emerge when input datasets came from the Short Trajectory MD with Analysis Floe.
Functionalities have been added to the NESGMXChimera Cube to detect the presence of protein tumbling restraint information in the Bound Input Dataset and apply them for bound state free energy calculations. This automatically propagates tumbling restraints in NES if they were requested and successfully applied in an upstream Floe that runs equilibrium MD.
The NESGMXChimera Cube is now also equipped to handle snapshots from multiple independent simulations of a ligand, as opposed to only working with a single simulation.
The NESGMXChimera Cube adds a new OEField called restraint_flags of type StringVec to the records and sends it downstream. If this field is not found in the record input to the NESGMX Cube, it will fail with error.
The NESGMX Cube switches to the Berendsen barostat if tumbling restraints are used in NES. In the absence of these restraints, the Parrinello-Rahman barostat is used.
v 6.3.6 April 2024
General Notice
A cost reduction switch has been added in the Nonequilibrium Switching Floe, a user switch option to reduce costs by restricting AWS GPU instances to only lower cost instances; it is On by default.
Minor cost optimization has been added to the Short Trajectory MD with Analysis and Induced-Fit Posing (Confined) Floes.
A binary star map option has been added in the Edge Mapper for RBFE calculations Floe.
Floe Updates
NES Floe titles have been updated:
“Equilibration and Non Equilibrium Switching” to Equilibration and Nonequilibrium Switching
“Non-Equilibrium Switching Recovery” to Nonequilibrium Switching Recovery
“Non-Equilibrium Switching” to Nonequilibrium Switching
The Edge Mapper for RBFE calculations Floe now supports the binary star map option, and the ligand hub(s) autoselection scheme based on the similarities to other ligands has been added.
The Nonequilibrium Switching and Equilibration and Nonequilibrium Switching Floes have been updated by adding a cost-switching flag.
In the NES Floes Equilibration and Nonequilibrium Switching and Nonequilibrium Switching, the recovery output dataset has been replaced by a record file to avoid overloading the Orion Database. In case of issues, the file can be input to the Nonequilibrium Switching Recovery Floe trying to recover the affinity results.
New Cubes
The NES Cost Switch Cube enables the selection of the cost-switching flag for NES.
An NES Gromacs Cost Saving Cube is optimized to run on selected AWS instances.
Cube Updates
The B-factor Threshold in Percentage parameter has been added to the Docking Checker Cube and the IFP Report Info Gather Cube, to determine the low B-factor binding site residues to evaluate when comparing with the target receptor and computing the fluctuation of the active site during the MD simulation, respectively.
The Gromacs Chimeric Cube has been updated to improve the fidelity of alchemical chimeras for polycyclic compounds involving changes affecting which atoms are common to two or more rings. For NES edges affected by this bug fix, users should expect greater computational stability (i.e., fewer switch crashes and no failed edges) and more accurate relative binding free energy predictions.
In the NES Floes Equilibration and Nonequilibrium Switching and Nonequilibrium Switching, the Recovery Output Dataset Writer Cube has been replaced by a Record to Record File Converter Cube.
In the Nonequilibrium Switching Recovery Floe, the Recovery Dataset Reader Cube has been replaced by a Record File to Record Converter Cube.
v 6.2.1 February 2024
General Notice
OpenMM 8.0 and Gromacs 2023.3 support has been added to the Floes.
Some major external package dependencies have been updated, including
numpy,scipy, and the CUDA toolkit.Bespoke and custom force fields can now be applied to simulation flasks. A bespoke force field is specific to certain ligands or parameters, such as torsion angles, and will override the associated parameters from a general or custom force field. They can be loaded using the Associate Force Field Offxml Files To Ligand Record Floe. A custom force field is a comprehensive force field (presumably with optimized parameters) and will be applied to the entire system when Ligand Force Field in the Force Field Cube is set to
Customand the corresponding custom force field files are uploaded. When providing a custom force field, the general force field will not be used at all.It is now possible to generate smaller simulation flasks by using a rectangular box and applying gentle restraints to the stable portion of a protein to limit protein reorientation. Selection of the stable portion of the protein involved in protein tumbling restraints is automated. This approach can also be used to stabilize a protein for other purposes, such as stabilizing a membrane protein in a water box without including the membrane.
v6.2.1 pins
orionmdcore==2.3, which includes new features and bug fixes, including a ligand Elf10 partial charge assignment bug fix. Detailed release notes of version 2.3 can be found here: Molecular Dynamics Core Package Release Notes.The Floe Compare Experimental Affinity with Nes Results has been renamed to NES Result Utilities.
New Floes
The Associate Force Field Offxml Files With Ligand Record Floe associates bespoke force field files content with the ligand dataset, producing an output dataset that serves as input for subsequent MD Floes calculations. When the bespoke force field content is identified in the dataset, the bespoke force field will be applied to the system if the prefer_bespoke switch in the Force Field Cube is turned On. The prefer_bespoke switch is On by default.
Floe Updates
The Induced-Fit Posing (Confined) Floe now supports SMI format for ligand input.
The Edge Mapper for RBFE calculations Floe now supports a star map with a user-selected hub, with the required input of the name of the hub (reference) ligand which will be used as a center ligand of the star map.
The Floe Compare Experimental Affinity with Nes Results has been renamed to NES Result Utilities
New Cubes
The SMIRNOFF Force Field Files Loader Cube loads SMIRNOFF force field files and associates their contents to the dataset, generating an output dataset.
The Specify Protein Tumbling Restraints Cube identifies the stable region of a protein, excluding the binding site, and saves the corresponding restraints information into the dataset.
The Bound Protein Ligand MD Health Checker Cube checks if there is restraint information saved on the record when the solvent box is rectangular. If there are no restraints applied to the system and a rectangular box has been selected, the job will be stopped to avoid unwanted interactions between the solute and its periodic images.
Cube Updates
Edges involving stereocenter inversion are now less likely to fail during NES runs. Since the introduction of proper stereo handling in version 6.1.2, some users have encountered warning messages of the form “MCS remapping warning: expected N matching atoms after remapping, but got M.” Previously, these warnings may or may not have been associated with an edge failure. In this release, the code has been improved and, though a similar warning may still be emitted, it is expected to be generally benign, and related edge failures should be reduced or eliminated. To assist with issue designation, the relevant message for all such issues is changed to “MCS v2 remapping warning: …” as of version 6.2.1.
In the OEMapper Cube Cube, a string parameter, map_type with the options “Default map” and “Star map (with user-selected hub)”, has been added.
v 6.1.7 September 2023
General Notice
We have updated some package dependencies:
orionmdcore==2.1.2.Openeye-orionplatform==6.0.0.Openeye-snowball==0.26.1.
v 6.1.4 September 2023
General Notice
In this release we have mainly focused on minor bug fixing.
Floe Updates
Search keywords have been added to the Induced-Fit Posing (IFP) Floes and the Short Trajectory MD with Analysis Floe.
In the Induced-Fit Posing (Confined) Floe, site residue matching between proteins for the site similarity measures has been improved.
The Bound Protein-Ligand MD, Short Trajectory MD with Analysis, Solvate and Run MD Floes now use the parallel iteration checker Cube to enable the scalability of the Floes.
Cube Updates
The Gromacs Run Cube has been updated to have better handling of GROMACS warning message control.
v 6.1.2 July 2023
General Notice
The Gromacs MD engine to run MD and Free Energy has been updated to the 2022.3 release. This update should have better MD performance in addition to bug fixing.
In NES, edges involving stereo center inversion are now supported. Specifically, NES edges involving changes in chirality (both R/S inversion and achiral/chiral site introduction) are now implemented correctly. Prior to this bug fix, directed edges with R->S chiral inversion were implemented as R->R and the reverse S->R transitions were implemented as S->S. This previous behavior led edges connecting two enantiomers to incorrectly estimate relative binding free energies as approximately 0 kcal/mol. Moreover, prior to this bug fix, directed edges representing achiral->chiral conversions may or may not have introduced the intended chirality (though chiral->achiral transitions were correct).
Floes for Induced-Fit Posing (IFP), a three-step protocol for the correct prediction of the binding pose of a ligand that requires induced-fit of receptor binding site residue side chains, have been added.
New Floes
Induced-Fit Posing (Confined) This Floe performs an IFP three-step protocol. The How-to Guide and tutorials can be found here.
Induced-Fit Posing (IFP) Prep: Pruning reference receptor for docking This Floe prunes the side chains in the binding site of the reference receptor for docking. The output datasets can be used as an “Optional Pruned Reference Receptor Input Dataset” in the “Optional Inputs” section of the Induced-Fit Posing (Confined) Floe.
Compare IFP Results to Target Receptor This Floe performs the comparison of the IFP results with the target receptor bound to the docking ligand and generates a Floe report.
Floe Updates
The Edge Mapper for RBFE calculations has been patched to remove dangling nodes not involved in cycles
The Bound Protein-Ligand MD, Short Trajectory MD with Analysis, Solvate and Run MD Floes now run MD simulations in chunks by using a Floe cycle topology to overcome the AWS session limit and make long MD simulations possible in Orion.
New Cubes
MutatedInputChecker: This Cube checks if an optional pruned reference receptor is given.
ActiveSiteDetectCube: This Cube finds an active site from the MDComponents object and saves a list of the active site atom indices.
MutateActiveSiteCube: This Cube prunes receptor binding site side chains to ALA to allow more space in the binding site, avoiding heavy clashes during docking. The binding site is defined based on the cut-off distance from the cognate ligand of the input reference receptor.
SetMapIdxCube: This Cube sets the reaction map index property of each atom of the primary molecule to its original atom index.
ResetAtomIdxCube: This Cube sets the AtomIdx of each atom of the primary molecule to MapIdx.
AdditionalPOSITSwitch This Cube switches whether to run an additional POSIT with an additional OEDocking method or not.
IFPPositCube: This Cube generates relaxed high probability poses for ligands that are analogs of the ligand in the receptor. This cube is suitable for pose generation when working with multiple receptors.
POSITGatheringCube: This Cube creates one record per ligand for post-processing. The primary molecule of the per-ligand record is the multi-conf mol of the Posit poses.
AddReceptorTypeCube: This Cube sets the receptor type field to the record.
PoseMMPBSACube: This Cube computes the MMPBSA score and BintScore from the last frame of the MD stage.
MinPoseGatheringCube: This Cube creates one record per ligand for post-processing. The primary molecule of the per-ligand record is the multi-conf mol of the minimized poses.
ClusterCube: This Cube runs clustering for the deduplication of the poses.
DockingCheckerCube: Given a target receptor to compare, this Cube checks the goodness of the posing results.
IFPIDSettingCube: This Cube sets the integer ID for each simulation flask as well as a descriptive title string. If the input molecule on a record has multiple conformers, these are split into singles, each with its own ID. If a complex will be formed, this Cube should be used on ligands before forming the complex.
IFDMinimizeCube: This Cube performs multi-layered restrained energy minimization on the docked pose/reference receptor conformation.
ComputeScoreCube: This Cube computes the IFP score.
ComputeInitScoreCube: This Cube computes the step 1 IFP score.
IFPBintScoreInitialPoseAndTrajectory This Cube computes the BintScore and iBintScore over the selected frames of the induced-fit trajectory.
IFPConcatenateTrajBintScoreCube: In this Cube, induced-fit trajectory BintScores and iBintScore are concatenated based on preexisting per-conformer vectors.
IFPClusterPopAnalysis This Cube analyzes the induced-fit trajectory clusters in terms of their occurrence, size, and proximity to the starting pose(s).
IFDOutputPrepCube: This Cube takes the last coordinates from the MDDataRecord and saves the updated DU in the “InducedFitPoseDU” field and updates the primary molecule coordinates.
IFPRankSetting This Cube sets the IFP Rank field and resets the primary molecule title with the suffix “_IFP_rankN”.
IFPOrderOutputRecords This Cube orders the records of the output dataset based on the IFP Rank field.
ExtractClusAvgFromTraj This Cube extracts a main cluster average from the MD trajectory analysis results.
PLAnalysisInputCheckerCube: This Cube checks if the input dataset is the output dataset of MD Analysis Floes. If True, it emits the input to the failure port.
SolvateMDInputCheckerCube: This Cube checks if the input dataset is the output dataset of MD Floes. If True, it emits the input to the failure port.
Cube Updates
The Gromacs Chimeric Cube has been updated to build a per-frame chimeric molecule. In addition, edges involving stereo center inversions are now correctly handled.
The Gromacs Run Cube has been updated to support the 2022.3 Gromacs release.
v 6.0.3 April 2023
General Notice
The MD API Dataset Converter Floe and Plain Molecular Dynamics Floe have been updated.
Floe Updates
The MD API Dataset Converter Floe has been updated to support the conversion from datasets serialized by using pickle to new datasets, where the serialization/deserialization is handled by using JSON. The MD Floes have been updated to automatically perform the dataset conversion, and no action is required by the users. However, dataset conversion is required for old datasets produced by custom customer Floes.
The Plain Molecular Dynamics Floe, which runs MD simulations in chunks by using a Floe cycle topology to overcome the AWS session limit and make long MD simulations possible in Orion, no longer pins instance type and spot policy. Users can now choose cheaper and faster instances for long MD simulations.
New Cubes
Concatenate Trajectory BintScore Components Cube. This Cube concatenates protein-ligand trajectory BintScores based on preexisting per-conformer vectors.
Cube Updates
The RBFECEdgeGathering Cube has been updated to support old mapper datasets.
v 5.6.1 December 2022
General Notice
Improved Edge Mapper.
Dual-trajectory ensemble MMPBSA support in the Short Trajectory MD with Analysis Floe.
Floe Updates
The Edge Mapper for RBFE calculations Floe has two improvements:
Alchemical changes between ligands of different formal charges are now allowed. No charge-correction is attempted along the edge at this time; this will change soon.
Edge scores are calculated for user provided mapper files.
The Short Trajectory MD with Analysis Floe now computes both the single-trajectory and dual-trajectory ensemble MMPBSA scores during the trajectory analysis.
New Cubes
2-traj MMPBSA Cube. This Cube computes the dual-trajectory ensemble MMPBSA.
Cube Updates
The OE Mapper Cube has been improved. Now the user provided mapper files are displayed with the calculated edge scores. In addition, edges between ligands with different charges are now allowed although no charge corrections are attempted at this stage during the NES switching. Our testing shows the uncorrected results are still reasonable though we will put in the correction soon.
The Affinity Plot Cube has been updated to add the correlation plot of dual-trajectory ensemble MMPBSA vs. experimental data to the existing single-trajectory ensemble MMPBSA and BintScore correlations vs experiment.
v 5.5.3 September 2022
General Notice
In this release we have mainly focused on bug fixing. Improvements have been done of the Chimera MCSS calculation, ligand isomeric SMILES matching, and Chimeric molecule frame alignment.
Cube Updates
The RBFECEdgeGathering Cube has been improved for better handling of ligand matching, by using isomeric SMILES.
In the NESGMXChimera Cube, the chimera molecule frame alignment, to start the NES switching, has been improved.
Compatibility Notice
Mapper Datasets generated prior to the MD Affinity package version 5.0.5 are no longer supported by the NES Floes, and they must be recreated by using the Edge Mapper for RBFE calculations Floe
v 5.5.0 July 2022
General Notice
In this release the major change is the harmonization of the Short Trajectory MD with Analysis (STMD) Floe with Nonequilibrium Switching (NES). Before this, after running STMD, for NES another equilibration run had to be made to get both ligand bound and unbound simulations of adequate length (6ns) for NES. Now this second equilibration run is unnecessary: STMD now runs both both ligand bound and unbound simulations of 6ns by default, producing output datasets ready for input to NES. Plus, you have the trajectory analysis available for those same equilibrium runs. This 6ns default trajectory for STMD is longer and therefore more expensive than the original 2ns run but we think it is an overall savings by not running a whole extra equilibration step for NES. It is easy to simply reset the new default 6ns back to 2ns if you want to have the faster, cheaper STMD run as before.
Additionally:
A Freeform analysis of the unbound state has been added to STMD.
A new formulation of the BAR overlap metric for the NES work distributions is incorporated into NES edge Floe reports
G4/G5 AWS instances are now supported for OpenMM/GROMACS
All Floes have a new brief parameter and Floe categories.
New Floes
Compare Experimental Affinity with End Point Analysis Results: This Floe is for comparing the short trajectory MD analysis results (binding affinities predicted using the MMPBSA and BintScore methods) with experimental data.
Floe Updates
Short Trajectory MD with Analysis (STMD) has been updated to harmonize with Nonequilibrium Switching (NES). STMD now runs both both ligand bound and unbound simulations of 6ns by default. Additionally, freeform is now run on the unbound ligand, producing a separate dedicated dataset.
The NES Floes have been updated with a new edge graph Floe report to show the predicted absolute and relative binding affinities. The BAR overlap metric has been added to the NES Floe reports.
The Solvate and Run MD Floe is updated to support apo protein simulations.
New Cubes
Bound and UnBound Splitting Cube. This Cube emits the primary molecule through the bound port and the standard out port to launch separated ligand-bound/unbound simulations.
Unbound FreeForm Cube. This Cube estimates the Gibbs free energy to convert an ensemble of solution conformations into a state where only a single specific conformation is present, and creates XY plot and table image of free energies of conformer ensemble.
Unbound Ligand Preparation Cube. This Cube checks whether the primary molecule (ligand) has a single conformer, and reset the title.
FreeForm Output Ligand Setting Cube. This Cube reads a Freeform record, chooses starting conformations for unbound MD simulations. The conformations are selected based on the probabilities of state associated with each conformation, and users can choose to ignore the Freeform results and use the bio-active conformation instead.
Unbound Traj to OEMol Cube. This Cube converts unbound MD trajectories into a multi-conformer ligand OEMol.
Unbound Traj MMPBSA Cube. This Cube computes molecular mechanics potential energies and solvation energies of the unbound MD trajectory snapshots. The Poisson-Boltzmann and Surface Area methods in the OEZap toolkits are used in the solvation energy estimation. The computed energies in the unit of kcal/mol are attached to the record as per-frame vectors of floats.
Unbound MD Conformer Gathering Data Cube. This Cube gathers conformer records of a ligand and emits a new single record per ligand. The assembly of the conformer records is attached to the record as a record vector.
Unbound Conf Trajs To Ligand Traj Cube. This Cube combines individual conformer trajectory OEMols into single multi-conformer ligand OEMol, in preparation of clustering.
Unbound Cluster Ligand Traj OEMol Cube. This Cube clusters unbound ligand multiconf MD trajectory OEMol.
Ligand Cluster Selection Cube. This Cube selects frames from the clustering analysis.
EdgeFloeReportCube. This Cube generates an edge graph Floe report for the relative and absolute binding affinities.
Cube Updates
The OEMapper Cube can also use clustering info to generate the map edges.
The NESAnalysis Cube is now calculating the BAR overlap metric for the work distributions.
v 5.0.5 April 2022
New Floes
None
Floe Updates
The Equilibrium and Nonequilibrium switching Floe now generates a tarball file of key results for download.
The Compare Experimental Affinity with NES Results Floe now generates a tarball file of key results for download.
The NES Recovery Floe now generates a tarball file of key results for download.
The Compare Experimental Affinity with NES Results Floe can now aggregate multiple input datasets to enable users to include the results from previous NES runs.
All the Short Trajectory MD Floes are by default pinning g3 spot instances for reliability.
All the NES Floes are pinning r5.4xlarge and r5.2xlarge Chimera cube instances for better Gromacs compatibility.
MD Analysis Floes are now able to handle any user specified ligand titles, including duplicate ligand titles.
In MD Analysis Floes, generating the results tarball file is now optional.
New Cubes
DDGAggregator cube. The Aggregator cube aggregates the relative binding affinity results produced by Nonequilibrium Switching runs.
Cube Updates
The OE Mapper cube is now sorting the edges by isomeric SMILES.
The NES Analysis cube is now setting the output units.
v 5.0.3 December 2021
General Notice
Edge Mapper for RBFE calculations
Long Trajectory MD supports
OpenFF 2.0 support
Improved Implicit solvent MD
Refactoring
Bug Fixing
New Floes
Edge Mapper for Relative Binding Free Energies. A starting edge mapper for RBFE calculations mainly based on LOMAP with a new ROCS similarity heuristic
Long MD Simulations. Performs long MD simulations starting from the output of the Protein-Ligand or the Solvate and Run MD Floes. The Long MD runs in chunks of max 10 hrs to overcome AWS parallel cube running time limitation
MD API Dataset Converter. The old openmm_orion MD pkg have been refactored in multiple pkgs (orionmdcore and oemdaffinity). As consequence a change in the API has happened. The MD API Datatset Converter Floe allows to convert datasets generated from the old openmm_orion pkg version <=4.0.2 to the new API >=5.0.3
Floe Updates
The MD Floes and Analysis have now better support for Implicit Solvent Calculations.
Nonequilibrium switching Floes are now uploading file results.
New Cubes
RBFECMapper cube. The mapper cube produces edges made of ligand pairs to run relative binding affinity calculations starting from a given set of ligands
RBFECMapperFloeReport. This cube is designed to work with the mapper cube output to generates the Floe report for the Orion UI displaying the graph of the generated edge map network
RBFECMapperResultToFile. This cube uploads a file to Orion with the mapper cube results
MDProxyCube cube.The MD Proxy cube runs chucks of MD simulations in cycles till the desired total md running time has been reached
MDRecoveryRestartProxyCube. This cube is able to restart or recover a previously run long MD simulation and it is used as support of the MD proxy cube.
Cube Updates
All the Protein_Ligand Analysis cubes have been improved to support Implicit Solvent
TrajToOEMolCube cube. Performance improvements and long md trajectory support. The frame stride parameter has been added to the list of parameters
v 4.0.1 June 2021
General Notice
Nonequilibrium Switching floes for Relative Binding Free Energy (RBFE) affinity calculations
New knowledge-based ensemble score BintScore for pose stability
Support for taking Posit multi-receptor output as input to protein-ligand MD
OpenFF 1.3.1a1 support
Bug Fixing
New Floes
Nonequilibrium Switching. Relative Binding Free energy calculations and estimate of Binding Affinity, starting from equilibrium MD calculations of the bound and unbound ligands.
Ligand Bound and Unbound Equilibration for NES. Performs the equilibrium MD calculations of the bound and unbound ligands needed for NES, starting from an input protein and a set of posed ligands.
Equilibrium and Nonequilibrium Switching. Equilibrium MD runs followed by Nonequilibrium Switching, starting from an input protein and a set of posed ligands.
Nonequilibrium Switching Recovery. This floe attempts to produce results from uncompleted NES runs, starting from the recovery file.
Compare Experimental Affinity with NES Results. Datasets from a completed NES run can be re-analyzed by comparison to a new set of experimental data.
Floe Updates
Short Trajectory MD with Analysis. The Bint score analysis has been added and Posit support
Analyze Protein-Ligand MD. The Bint score analysis has been added
Cofactors have been added to the MMPBSA score calculations
New Cubes
BoundUnboundSwitching cube. This cube accept a flask and emit on the bound port the flask containing the a complex while the unbound flask is emitted on the standard output port
RBFECEdgeGathering cube. This cube gathers the MD equilibrium simulation data related to the Bound and Unbound simulations and creates edges used to perform relative binding affinity calculations
NESGMXChimera cube. This cube generates a chimera molecule and injecting it on select frames from the Bound and Unbound simulations for the Gromacs forward and reverse Nonequilibrium simulations
NESGMX cube.This cube runs the forward and reverse Bound and Unbound starting equilibrium frames by using Gromacs
NESAnalysis cube. This cube collects the data generated from the Nonequilibrium runs and calculates the relative binding affinities
PlotNESResults cube. This cube generates plots for the Relative binding affinities and the estimated Absolute Affinities versus the provided experimental data. Relevant statistics are calculated and tabled
PredictDGFromDDG cube. This cube applies the Maximum likelihood Estimator method to predict the absolute binding affinities starting from the relative binding affinities
BintScoreInitialPoseAndTrajectory cube. This cube calculates a trajectory ensemble BintScore relative to an initial pose.
Cube Updates
Complex Preparation Cube. This cube has been updated to give better support for clashes detected between the ligands and the other flask components for Posit support
v3.0.1 November 2020
General Notice
OpenFF 1.3.0 and 1.2.1 support
Bug Fixing
v2.0.0 September 2020
General Notice
Ligand centric cluster Analysis
New Parametrization Engine
OpenFF 1.2.0 (Parsley) support
Bug Fixing
New Floes
Solvate and Run Protein-Ligand MD. Runs MD on a Protein-Ligand system
Analyze Protein-Ligand MD. Performs Energetic and Clustering Analysis on the output produced by the
Solvate and Run Protein-Ligand MDFloeExtract Short Trajectory MD results for Download. Extract the results produced by the
Short Trajectory MD with AnalysisFloe as file to download
Floe Updates
A new Equilibration IV stage has been added to the Short Trajectory MD Floe
New Cubes
A new
MD Componentscube has been added to support the new Parametrization Engine
Cube Updates
None
v1.0.0
General Notice
Support for OpenFF 1.1.0 (Parsley), the March 2020 release from the Open Force Field Initiative
Support for Cuda 10.0
Exposed a new parameter in the
TrajToOEMol Cubeto set the water cutoff distance. This distance is used to select water molecules around the protein-ligand binding site for each trajectory frame and producing a multi conformer water moleculeImproved Documentation
New Floes
None
Floe Updates
New default parameters have been set on the STMDA Floe
New Cubes
None
Cube Updates
None
v0.9.6
General Notice
Support for the two force fields from the Open Force Field Initiative: Smirnof99Frosst and OpenFF 1.0.0 (parsley)
A new automatic color style is applied to the clustering in the
Short trajectory MD analysisto match the cluster colors in the Floe ReportFixed a wrong setting in the
Solvation Cubethat was placing solvent molecules too close to the solute. This could have produced un-realistic results for some systems where water molecules could have been placed inside proteinsFixed a bug in the GAFF/GAFF2 force field where 1-4 interactions were not correctly scaled
Fixed a bug related to un-wanted ligand atom re-ordering
Fixed a bug in the protein-ligand active site depiction
New Floes
A new Plain Gromacs Floe has been added to perform MD by using Gromacs .tpr files
Floe Updates
The Yank solvation free energy Floe has been removed
New Cubes
None
Cube Updates
The force field parametrization cube now support open force field 1.0.0 (parsley)
The trajectory to multi conformer cube is now adding to the record protein-ligand binding site close waters. These are used to perform MMPBSA calculations with explicit water
v0.9.4
General Notice
Upgrades to the trajectory analysis in Floe
Short Trajectory MD with Analysisexpose key results to the Analyze page and 3D visualization page in Orion.Fixed bug in setting restraints in GROMACS cubes.
Fixed bug in ordering results in the Floe report in Floe
Short Trajectory MD with Analysis.
New Floes
The MD Spruce Prep Floe has been removed. Proteins must be prepared with the Spruce Prep Floes available in the Classic Floes now.
The calculation of MMPBSA can now be also performed by using explicit waters (still experimental)
Floe Updates
The MD Spruce Prep Floe has been removed
The Simple MD Floe has been renamed the Plain MD Floe
New Cubes
A new cube has been developed to check the record size before writing to the Orion backend to avoiding Floe failures. The new cube has been added to all the Floes for sanity check.
A new cube to estimate the water number around a ligand-protein complex has been developed. The cube is used in the MMPBSA calculation with the explicit water flag set on
Cube Updates
Exposed MMPBSA ensemble average and standard deviation in the TrajPBSACube so that it can be displayed in the Analyze page in Orion.
MDTrajAnalysisClusterReport now generates trajectory average and median molecules for protein and ligand, with one conformer for each major cluster. These are exposed int the 3D visualization page in Orion.
ClusterOETrajCube now exposes a link to the per-ligand Floe report page so it is available in the Analyze page in Orion.
In ComplexPrepCube traditional references to the full periodic supermolecular ensemble as a “system” have been replaced with references to a “flask” by analogy with an assay well.
In MDFloeReportCube the Floe report now generates tiled links to individual ligands in the same order as the initial list of ligands.
In MDFloeReportCube the Floe report tiles now show how many major clusters were found for each ligand.
In MDNptCube and MDNvtCube the restraints are now correctly set in GROMACS for proteins consisting of multiple chains.
Hint interactions and Styles have been removed from receptors and ligands in the Protein, Ligand and FF parametrization setting cubes that could cause problems along the MD analysis stages (debugging is in progress)
A bug has been fixed in the ligand Elf10 charging cube that was causing problems when carboxylic acid was present in a ligand to be charged
The Trajectory to OEMol, Interaction Energies and PBSA calculation cubes have been updated to account for the explicit water in the new MMPBSA calculation
Legacy Release Notes
This package is built using
openeye-orionplatform >=5.1.1, <7,openeye-snowball >=0.26.0,openeye-toolkits >=2023.2.3,openff-toolkit >=0.14.4, <0.15, andparmed ==3.4.4.
Legacy cubes and API points have been removed to upgrade old data formats.
Temporary resources are now deleted immediately after use.
A bug has been fixed that could cause Gromacs to crash when multiple water residue names were present.
Relaxed OpenEye Snowball version constraint to allow versions >= 0.26.0 (upper bound removed).
Temporary resources are deleted immediately after use.
An issue has been fixed with enforcing atom ordering for Gromacs that caused the Solvation Cube to consume significantly more memory than expected.
An issue has been fixed where residues of nonprotein components were renumbered after simulations.
The Delete MD Recovery Cube has been updated to use a less memory-intense protocol.
Various aspects of Orion MD Core have been refactored and improved significantly in this version. Please note that some changes are unfortunately API-breaking, which may affect existing implementations. These changes are explicitly marked as (API-breaking) in the release notes below.
The Solvation Cube has been updated as follows:
The default salt concentration has been increased to 150 mM from 50 mM.
The box size for small molecule is now determined considering the largest small molecule conformation along Omega sampling.
A new
md_cutoff_distancecube parameter has been defined to ensure that the minimum image convention is always enforced along MD runs.Ions in solutions are now placed far from the solute.
A new
min_number_of_saltcube parameter has been defined to ensure that the selected minimum number of ions are always present in solution. This parameter has been introduced to ensure future charge corrections change ligand mutations along RBFE calculations.
(API-breaking) The Collection Setting Cube has been refactored to use a new parameter,
collection_fields, to specify which collection will be set on the input record. Additionally, a new string parameter,operation, has been introduced to define the action to perform on the collection. Supported values for operation are"open","close", and"create".
Support has been added for storing the bond orders in trajectories generated by the MD cubes using OpenMM as the engine. Bond orders can be accessed in output trajectory files as follows:
import mdtraj # NOTE: mdcore must be imported for bond orders to be recognized by mdtraj, # even if mdcore is not explicitly used import orionmdcore # load the trajectory from the last stage traj_file = md_record.get_state_trajectory() traj = mdtraj.load(traj_file) # check if bond orders are present for bond in traj.top.bonds: assert bond.order is not None
MDDataRecordhas been improved in the following ways:MDDataRecordnow internally uses the link, as opposed to integer IDs, to reference MD data stored as Orion resources (e.g., shards and files). ID fields from old records are automatically upgraded to links onceMDDataRecordis initialized.MDDataRecord.get_valueandMDDataRecord.set_valuenow return/expect Orion resources instead of links when dealing with link-type fields. For example:# generic record link_dict = record.get_value(MDFields.mddata) # returns a link dict referencing the shard link = APISession.get_link(link_dict) shard = link.get_shard() record.set_value(MDFields.mddata, link) # sets with a link referencing the shard # MDDataRecord md_record = MDDataRecord(record) shard = md_record.get_value(MDFields.mddata) # returns the shard md_record.set_value(MDFields.mddata, shard) # sets with the shard
Related utility functions and tools, including
mdocli, have been refactored to support links stored onMDDataRecord.A new function,
get_stage, has been added toMDDataRecordthat can be used to retrieve MD stages by either stage index or name.MDDataRecord.get_stagesand other MD stage getters now return anMDDataRecordinstead of a generic record.(API-breaking) The input parameter of
MDDataRecord.get_stage_by_name,MDDataRecord.has_stage_name, andMDDataRecord.delete_stage_by_namehas been renamed fromstg_nametonamefor improved clarity and simplicity. Additionally, the parameter can now be provided as either a positional or keyword argument. For example:stage = md_record.get_stage_by_name("production") # stage name as positional argument stage = md_record.get_stage_by_name(name="production") # stage name as keyword argument
(API-breaking) The input parameter of the several member functions of
MDDataRecordrelated to getting and setting the MD stage data has been changed such that the user can provide either the name or the index of the stage. The name of the parameter has been changed fromstg_nametostage_idto reflect this new capability. Additionally, the parameter can now be provided as either a positional or keyword argument. For example:traj_file = md_record.get_stage_trajectory("production") # stage name as positional argument traj_file = md_record.get_stage_trajectory(stage_id="production") # stage name as keyword argument traj_file = md_record.get_stage_trajectory(-1) # stage index as positional argument traj_file = md_record.get_stage_trajectory(stage_id=-1) # stage index as keyword argument
Atoms in all components except for ligand and other_ligands are now reordered in
MDComponentsto enforce and satisfy the requirements for GROMACS topology. This should allow systems that were previously failing in GROMACS to be successfully simulated.A new property,
designunit, has been added toMDComponentsfor converting and retrieving anOEDesignUnitobject fromMDComponents.
Package dependencies have been updated to include the latest versions.
The
use_gpu_cpuoption in theOpenMMSimulationsclass has been renamed to"use_gpu"and changed to take Boolean values.A
precisionoption has been added to theOpenMMSimulationsclass, allowing users to specify the numerical precision for simulations when using the GPU. Supported values for this option are"single","double","mixed", and"auto". Ifprecision="auto", the simulation defaults to mixed precision when available; otherwise, it falls back to single precision. The default value is"mixed", which requires mixed precision (i.e., fails when mixed precision is unavailable).
A rounding error has been fixed when calculating the total number of steps in MD runs.
An issue has been fixed that can lead to memory leaks when deserializing
MDComponentsfrom a record.A bug has been fixed in the Delete MD Recovery Cube that could cause a deadlock.
An issue has been fixed where no record is emitted from NVT/NPT cubes when the simulation runtime is zero.
Various smaller bugs have been fixed.
This patch adds support for the Sage 2.2.0 small molecule force field from Open Force Field.
The default Ligand Force Field is now “OpenFF_2.2.0”.
Improved input validation for the restraintWt parameter.
The CollectionSetting Cube now creates V2 ShardCollections by default. This behavior is controlled by the new V2 switch, which defaults to On.
Cubes that need to run OpenMM on a GPU now print a more informative error message when no GPU platform is available.
The IterChecker Cube no longer deletes the per-iteration MD stages (cycle_0, cycle_1, etc.) after concatenating them. This task has been delegated to the new DeleteMDStagesCube.
Cubes no longer attempt to load the reference positions when the Restraint to Reference switch is turned Off.
The package distribution now includes both the orionmdcore and oeommtools Python packages (oeommtools was released separately in a previous release).
The default value for the ForceFieldCube.ligand_forcefield parameter has been changed from “Gaff_1.81” to “OpenFF_2.0.0”.
The ForceFieldCube.ligand_forcefield parameter has a new “Custom” value. When this value is selected, the ligand force field is defined by inputting one or more SMIRNOFF XML files via the custom_offxml_file_in parameter.
When the ligand_forcefield is set to “Custom”, “OpenFF_1.3.1”, or “OpenFF_2.0.0”, and the new prefer_bespoke switch is turned On (the default), the force field can be augmented with bespoke parameters by providing them in the field specified by the bespoke_forcefield_field parameter. This field accepts a list of SMIRNOFF XML strings.
The new SolvationCube.random_seed_from_system_time parameter specifies whether Packmol should generate its random seed from the system time. The default (and historical) behavior is to use Packmol’s fixed default seed.
Residue numbers in the output topology now match those in the input design unit or molecule.
A bug in which the IterMDNptCube could schedule a cycle of zero length has been fixed.
This version of Orion MD Core has more flexible requirements:
>>> from importlib.metadata import metadata, requires
>>> metadata("openeye-orionmdcore")["Requires-Python"]
'>=3.9'
>>> requires("openeye-orionmdcore")
['click <9,>=8.1.3',
'h5py <4,>=3.1.0',
'lxml <5,>=4.6.0',
'mdtraj <2,>=1.9.6',
'numpy <2,>=1.21.2',
'oeommtools <2,>=1.2.1',
'openeye-orionplatform <7,>=5.1.1',
'openeye-snowball <0.28,>=0.26.0',
'openeye-toolkits >=2023.2.3',
'openff-toolkit <0.15,>=0.14.4',
'openmm <8.1,>=7.7.0',
'openmoltools ==0.8.8',
'parmed ==3.4.4',
'pdbfixer <2,>=1.8.1',
'pyparsing <3,>=2.4.7',
'tqdm <5,>=4.57.0']
Warning
This version reintroduces a bug, fixed in version 2.1.1, in which the
IterChecker cube incorrectly records the combined simulation time
as the time of the last stage. Furthermore, while version 2.1.1 requires
oeommtools==1.2.*, this version requires oeommtools==1.2.0.
Hence it is unable to leverage new features in oeommtools 1.2.1,
including the retention of residue numbers when converting from an OEMol
to an OpenMM topology. These issues are fixed in version 2.3.1.
Support has been added for systems containing iron and other metal cofactors, which previously failed during force field assignment.
A new cube type, Parallel Iteration Checker Cube was added to enable scalability of the (serial) Iteration Checker Cube.
This version v2.1.0 pins Python 3.9 and OpenMM 7.7.
Minor bug fixes have been made.
In the Solvation Cube, the “geometry” parameter name has been changed to “Periodic Box shape”. The parameter offers two options: a “cubic” or “rectangular” periodic box.
The Iterative NPT Cube has a better way of setting trajectory intervals for long simulations (>= 10 ns).
The Ligand Charge Cube now supports ELF10 charge assignment of macrocycles.
Since the first MD pkg release, we have been using Pickle to serialize and deserialize MD custom objects passed along the OpenEye data records. A main side effect of pickling them is that the class objects API is embedded inside the produced dataset. Therefore, updating the object APIs could generate issues. To overcome the problem, all the MD custom objects are now using JSON serialization and deserialization, which does not have this side effect. To avoid compatibility problems, in this release both serializers (pickle and JSON) are supported. However, the support for pickle will be dropped in future releases, and only the JSON serializer will be kept. The MDAPIDatasetConverterCube can be used to convert old datasets into new datasets where the JSON serializer/deserializer is used.
Iterative NPT Cube. This cube performs NPT MD simulations in the cyclic MD Floe (Plain MD Floe) on the provided system. If the MD cycle is complete, the cube emits the record to the “success” port and if not, it emits the record to the “cycle_out_port” port.
Iteration Checker Cube. This cube checks if the MD cycle is complete. If the MD cycle is complete, it emits the record to the “true” output port, and if not, it emits the record to the “false” output port.
The MDAPIDatasetConverterCube has been updated to support the conversion from old pickle datasets to the new JSON serializer/deserializer
The ComplexPrepCube has been updated to raise a fatal error if the protein cannot been found.
Add new Cube for checking the wall clock time of production runs
G4/G5 AWS instances support for OpenMM/GROMACS
Minor bug fixing
MDSpeedChecker Cube. This Cube estimates the wall clock time a production run will require and if the estimated time exceeds the 12 hour time limit, the Cube will fail the job.
All Cubes have been updated for improved exception handling.
New Cubes for Long Trajectory MD support; these are Cubes to enable iterative Floes.
Package dependencies update
Minor bug fixing
MDProxyCube. The MD Proxy Cube runs chunks of MD simulations in cycles till the desired total MD running time has been reached
MDRecoveryRestartProxyCube. This Cube is able to restart or recover a previously run long MD simulation. It is used in association with the MD proxy cube.
All the MD Cubes are by default pinning g3 spot instances to improve reliability.
Better Implicit Solvent Support
Add CPU support to OpenMM and Gromax
Add HMR support to Gromax
MDAPIDatasetConverterCube
All the MD cubes have been updated to support Implicit Solvent simulations
pKa Prediction Floes
v0.1.0 August 2025
General Notice
This package is built using
OpenEye-orionplatform==6.5.1,OpenEye-toolkits==2025.1.0, andOpenEye-Snowball==0.30.0.This is the first release of OpenEye Small Molecule pKa Prediction Floes package.
Legacy Release Notes
v0.1.2 September 2025
General Notice
This package is built using
OpenEye-toolkits==2025.1.1,OpenEye-orionplatform==6.7.0,OpenEye-Snowball==0.30.0, andOpenEye-orionmdcore==2.5.4.This is the first release of the OpenEye Structural Biology Floes package.