OEUniMolecularRxn
class OEUniMolecularRxn
The OEUniMolecularRxn class is designed to apply a chemical transformation to a molecule. The primary purpose of the class is to provide an efficient means of applying normalization reactions, although there are few restrictions on the types of chemical transformations that can be applied. The reaction used to initialize the class must contain only a single reactant. The provided transformation is applied iteratively until no further reactant patterns are matched. Caution must be used in defining the transformation as it is possible to provide a reaction that will cause a OEUniMolecularRxn object to iterate until memory is exhausted. For example, a reaction for which the reactant pattern matches the transformed product molecule will reapply the transformation indefinitely and must be avoided. Note, however, that atoms and bonds created by a transformation are excluded from involvement in iterative applications of the transformation.
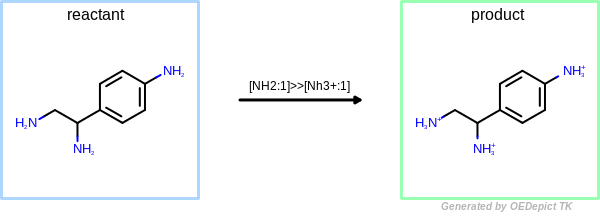
OEUniMolecularRxn
Example of amine nitrogen protonation using ‘OEUniMolecularRxn’. Transformations are applied to the starting material recursively
See also
Reactions chapter
OELibraryGen class
Example program UniMolRxn.cs
Constructors
OEUniMolecularRxn()
Default constructor.
OEUniMolecularRxn(const OEUniMolecularRxn &rhs)
Copy constructor.
OEUniMolecularRxn(const char * smirks, bool strictSmirks)
Creates a OEUniMolecularRxn object with a (SMIRKS) pattern.
Attempts to initialize an OEUniMolecularRxn
object with a reaction that has more than one reactant will
result in an initialization failure.
Initialization success can be tested using the operator
OEUniMolecularRxn.operator bool method.
The second argument to the constructor is used to specify the
interpretation of the SMIRKS semantics.
By default, the smirks string is interpreted using strict
semantics via a value true.
Passing a smirks value of false to the second method argument will relax
the strict SMIRKS restrictions.
A mapped reactant atom that does not have a corresponding mapped
product atom is valid, and will be destroyed as part of the
reaction.
Unmapped reactant atoms will be used to match the reactant
pattern, but are not destroyed when the reaction is applied.
The resulting SMIRKS like reactions may therefore be
more easily readable by humans as fewer atoms may be required to
be mapped.
See also
OEUniMolecularRxn.InitmethodSMIRKS definition describing the strict SMIRKS semantics
OEUniMolecularRxn(const char * smirks, const OEChem::OEUniMolecularRxnOptions &options=OEUniMolecularRxnOptions())
Creates a OEUniMolecularRxn object with a
(SMIRKS) pattern. The options class argument will be interrogated to determine the desired
strict smirks interpretation requested for the (SMIRKS) processing.
See also
OEUniMolecularRxn.InitmethodSMIRKS definition describing the strict SMIRKS semantics
operator=
OEUniMolecularRxn &operator=(const OEUniMolecularRxn &rhs)
operator()
bool operator()(OEMolBase &mol) const
Applies the transformation that the
OEUniMolecularRxn object contains on
the passed OEMolBase object.
Returns true if the reactant pattern matches at least one
time in the passed molecule.
If the OEUniMolecularRxn fails to apply any
transformations because the reactant pattern failed to match the
molecule, the method will return false.
operator bool
operator bool() const
Returns true, if the OEUniMolecularRxn
object was initialized with no failures using either the
constructors or one of the
OEUniMolecularRxn.Init methods.
The method will return false when the
OEUniMolecularRxn object has not been properly
initialized as it will be unable to apply transform operations
to molecules.
GetClearCoordinates
bool GetClearCoordinates() const
Returns whether the coordinates of a molecule are cleared before performing the chemical transformation.
See also
GetOptions
OEUniMolecularRxnOptions& GetOptions()
const OEUniMolecularRxnOptions& GetOptions() const
Returns the internal options class in the OEUniMolecularRxn
instance. A non-const instance supports changing the option control values for
options that are active during transformation application. Note that the
strict flag processing setting occurs when the SMIRKS or OEQMolBase
transformation is processed and not during application of the transformation.
See also
OEUniMolecularRxnOptions class
GetValidateKekule
bool GetValidateKekule() const
Returns whether the transformation validates the Kekulé form of the returned products.
See also
Product Kekulization section
Init
bool Init(const char *smirks, bool strict)
bool Init(const char *smirks, const OEUniMolecularRxnOptions &options=OEUniMolecularRxnOptions())
bool Init(const OEQMolBase &qmol, bool strict)
bool Init(const OEQMolBase &qmol, const OEUniMolecularRxnOptions &options=OEUniMolecularRxnOptions())
The first two method overloads initialize an OEUniMolecularRxn
object with a smirks pattern and either the explicitly provided strict flag
setting or via the strict setting from the provided options class.
The next two method overloads initialize an OEUniMolecularRxn
object with a OEQMolBase object and either the explicitly provided strict flag
setting or via the strict setting from the provided options class.
See also
SetClearCoordinates
bool SetClearCoordinates(bool clearcoords)Sets whether the coordinates of a molecule are cleared before performing the chemical transformation.
Note
The OEUniMolecularRxn modifies the molecular graph when applying the transformations. Generating valid 2D or 3D coordinates for the products is beyond the scope of the OEUniMolecularRxn class.
It is recommended to either clear the coordinates by invoking the
OEUniMolecularRxn.SetClearCoordinatesmethod or regenerate the coordinates of the molecule afterwards.If no transformation is applied the molecule will not be changed.
See also
SetValidateKekule
bool SetValidateKekule(bool validate)
Sets whether the generated products are simply returned after a Kekulization attempt, or if the Kekulé form is validated more rigorously. If the extra validation is enabled and an invalid Kekulé form is identified, alternatives are tried by adding and removing implicit hydrogens from aromatic nitrogens to produce a valid Kekulé form. No further attempt is made to return the most reasonable or chemically sensible result. By default, the extra Kekulé validation is enabled.
See also
Product Kekulization section