Spruce Prep Tutorial
The Spruce Protein Preparation Floe(s) use SPRUCE to prepare biomolecular systems for use in downstream modeling applications. The Floe generates a single or a set of prepared design units with associated depictions, e.g. to illustrate the protein-ligand binding site interactions, as well as the Iridium classification.
The preparation steps of the Floe include:
Expansion of the asymmetric unit to the biological unit (if necessary, e.g. if structure is from an X-ray crystallography experiment). The biological unit being what is known or believed to be the form the protein(s) take in-vivo or in-vitro.
Enumeration (default) or collapse of alternate locations.
Building missing pieces, e.g. partial sidechains, modeling missing loops and tails, capping chain breaks.
Placement and optimization of hydrogen atoms including tautomer enumeration of ligands and cofactors as well as evaluation of those tautomer states in the biomolecule structure.
The protein and ligand are assigned partial charges (AmberFF99SB and AM1BCC, respectively) and the output dataset is ready for downstream structure-based modeling tasks.
Floes Used in the Tutorials
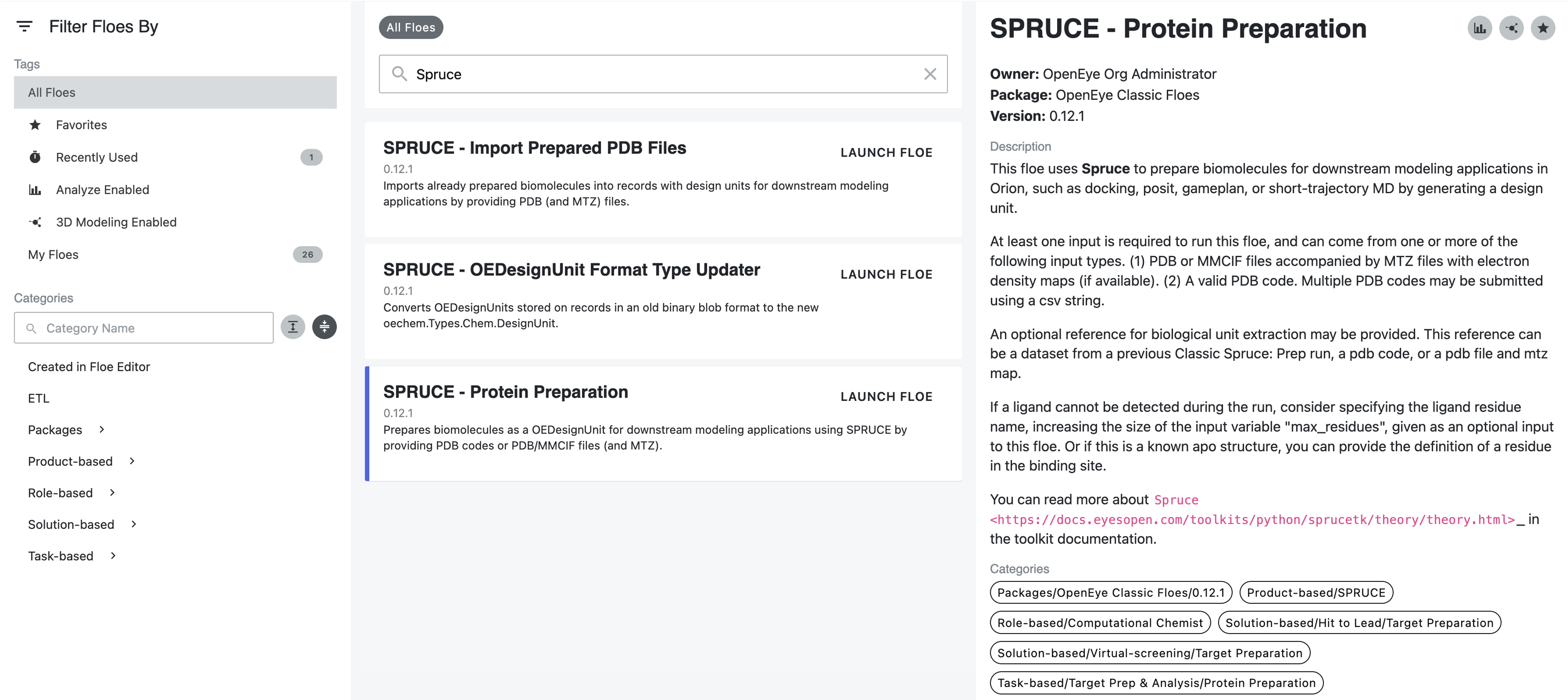
The floe used in this tutorial is documented here:
There are two primary methods for protein structure input, and both will be shown below. To run the Spruce protein preparation floe, navigate to the Floe tab in Orion, then:
Type “SPRUCE” in the search box.
Click the “SPRUCE - Protein Preparation” floe to open the Job Form.
Run the Spruce Protein Preparation Floe(s)
The first method for protein structure input takes a list of PDB codes and pulls the structures and their associated electron density maps (mtz files) from the PDB server hosted at the RCSB.
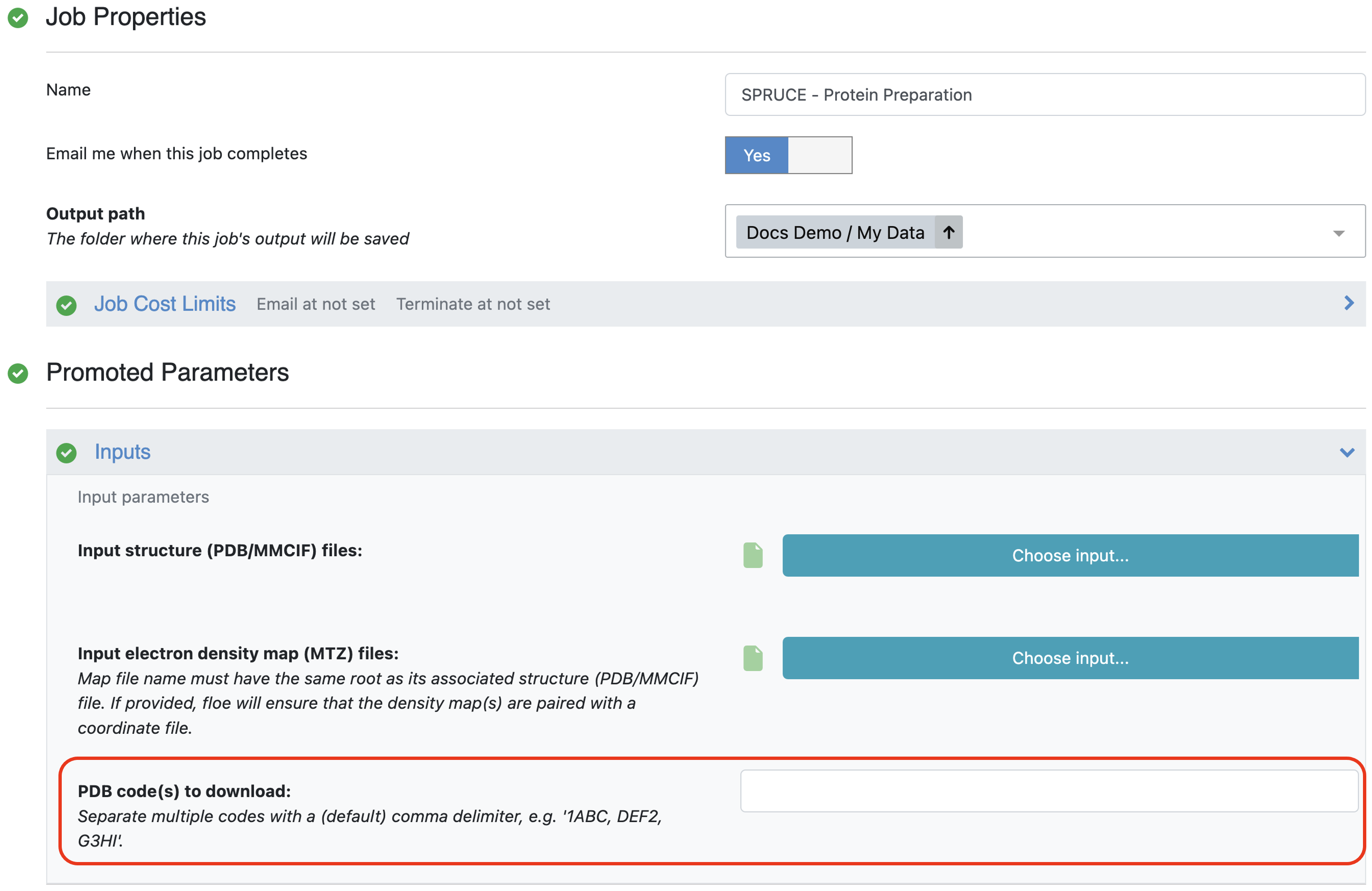
Provide PDB code(s) as input
The second method accepts a list of PDB files. These can already be uploaded to Orion or can be uploaded as part of the job submission. We recommend providing the associated MTZ files to enable calculation of the Iridium classification and to provide electron density grids for structure inspection after preparation.
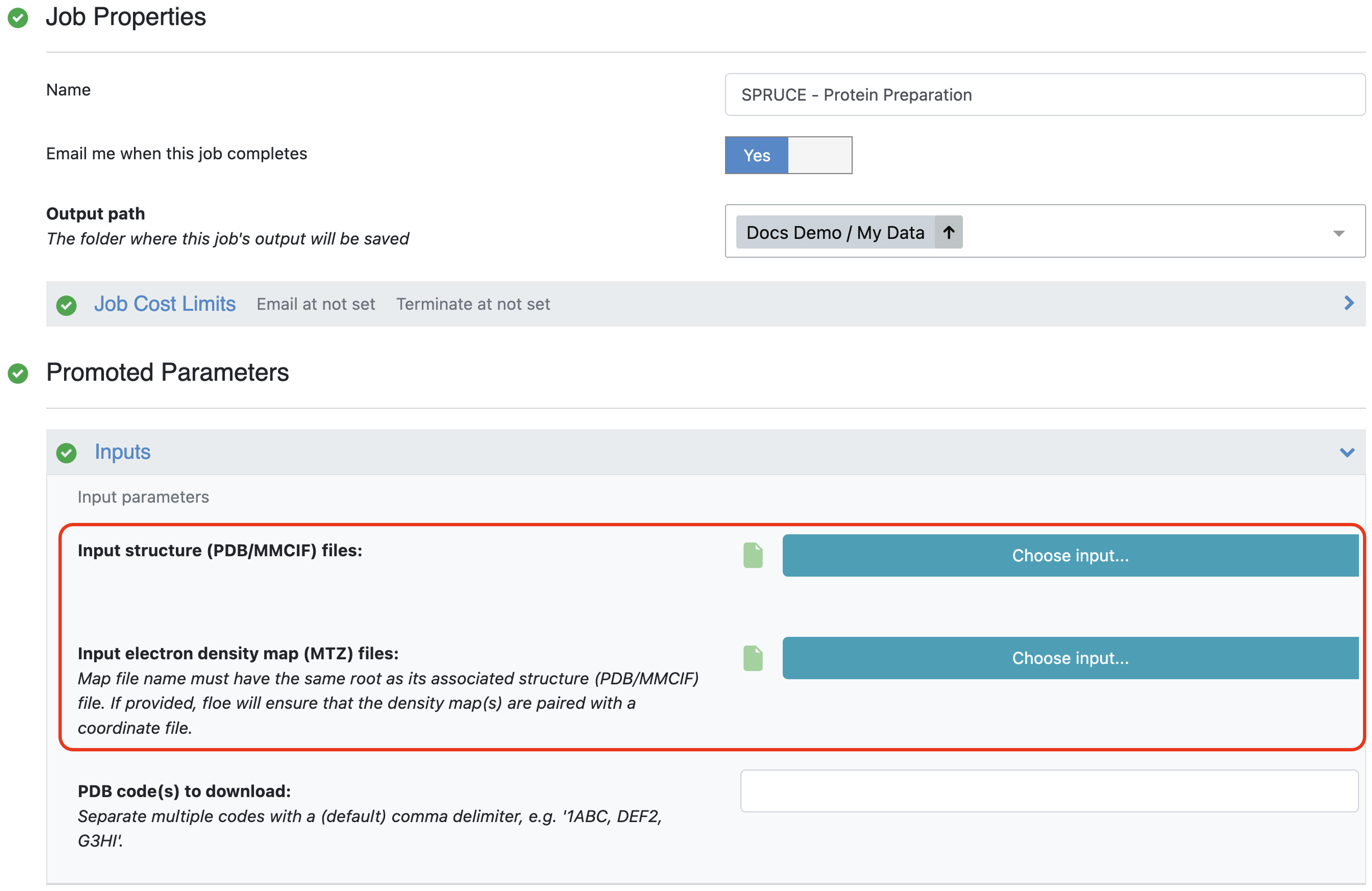
Provide PDB and MTZ file(s) as input
The files can be uploaded from a computer, pulled from a URL, or files already in Orion can be used.
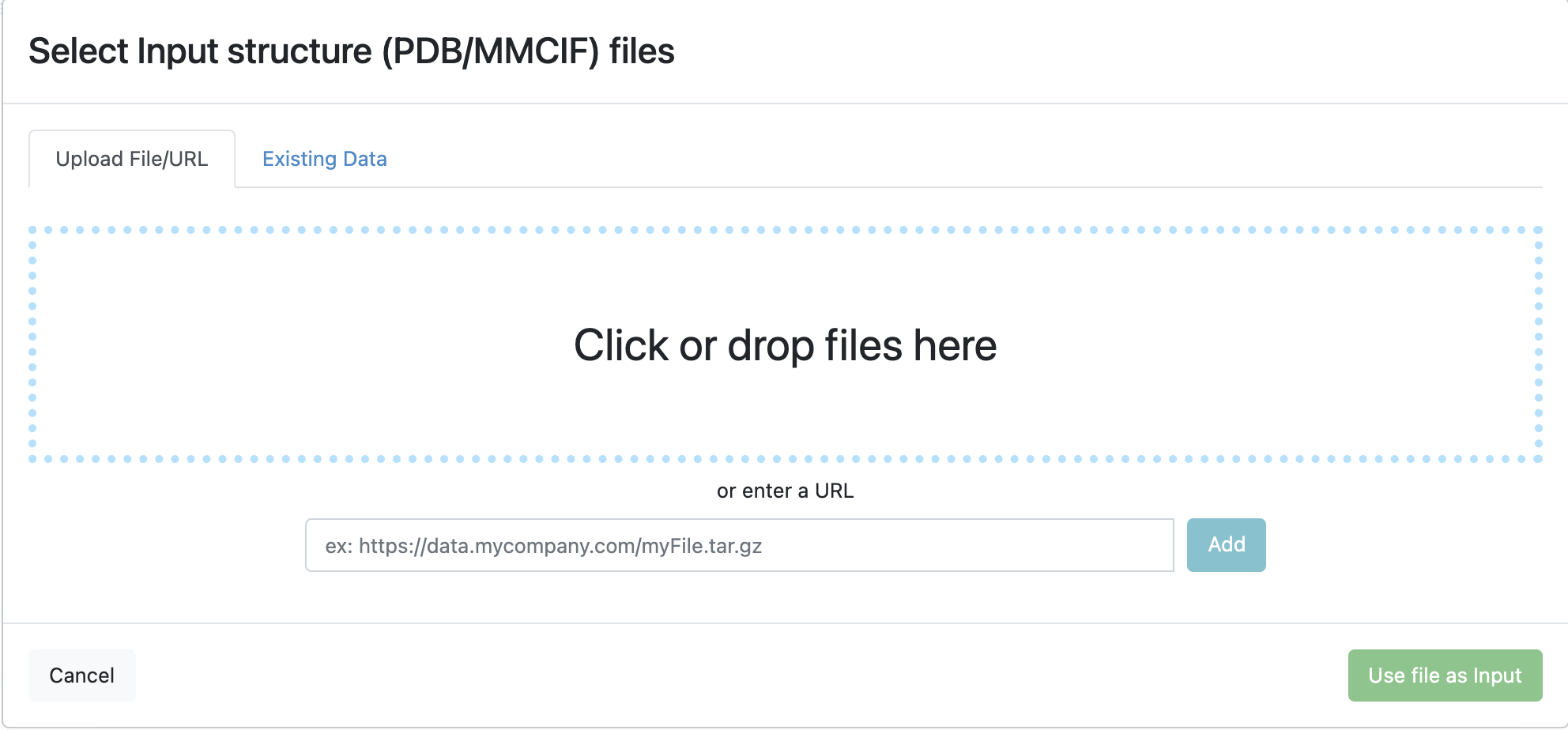
Files can be uploaded or existing files in Orion can be used
At least one protein structure must be input using either of the above described methods. The remaining inputs are optional and described in more detail in the FAQ and How To Guide.
Run the Protein Preparation Floes
An extracellular signal-regulated kinase 1/2 inhibitor (CDK2) is used in this tutorial to show how Spruce preparation can be done with or without a reference protein. When a reference structure is used, all submitted structures will be superposed, and the output design units will mirror the reference’s receptor. If no reference is used, all liganded design units will be output. Finally, if an apo structure is detected and no reference is given, pocket finding will generate design units based on structural pockets on the protein’s surface.
The most simple use is shown below using an experimental CDK2 structure (PDB code 5K4J) with only the
PDB code needed to run the
SPRUCE - Protein Preparation Floe.
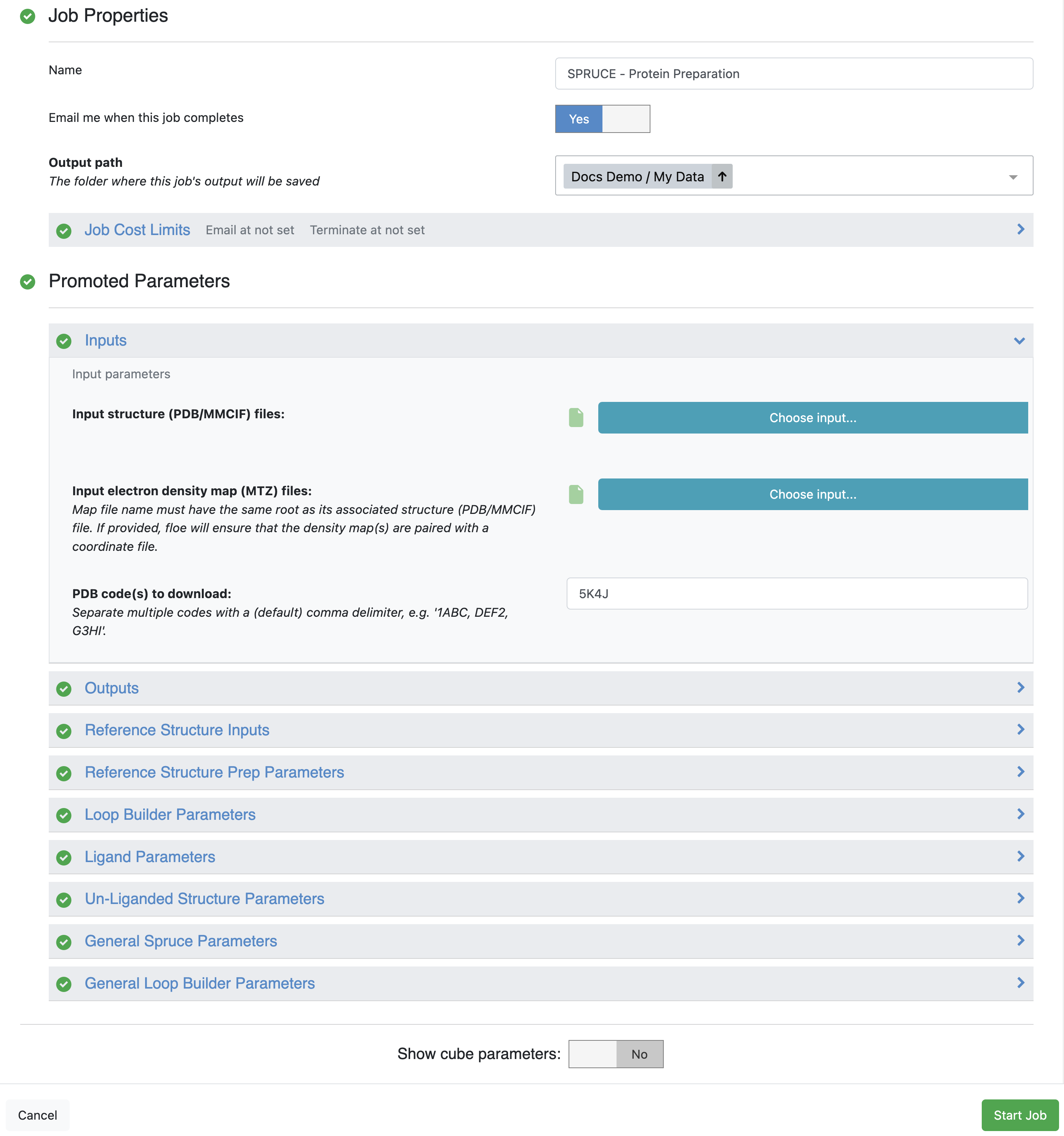
Spruce Floe Parameters
The output dataset can be viewed in the analyze page or the 3D view.
Run the Protein Preparation Floes Using a Reference Design Unit
Preparing with a reference structure begins similar to preparation without a reference structure. First the protein structure to be prepared are input using any combination of the previously described input methods. For this example we are going to prepare many related CDK2 proteins at the same time. The 5K4J structure is based on the P24941 UniprotKB ID, we can use this ID to search the PDB:
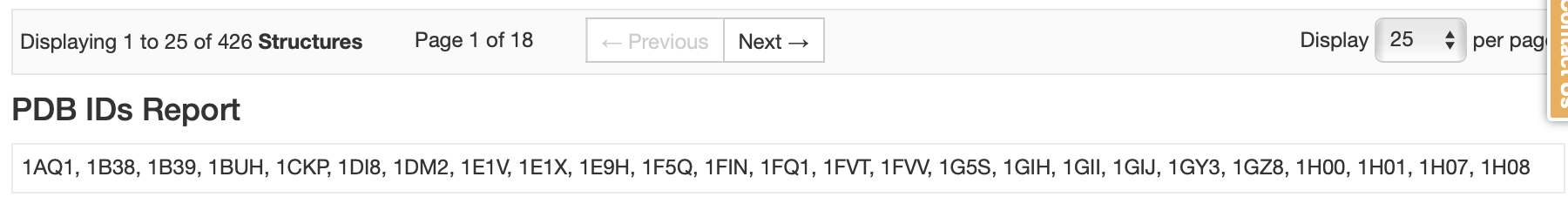
RCSB Search Results
This results large list of PDB codes (with Tabular Report showing Entry IDs). For this tutorial, we are going to prepare the first 25 hits with Spruce (1AQ1, 1B38, 1B39, 1BUH, 1CKP, 1DI8, 1DM2, 1E1V, 1E1X, 1E9H, 1F5Q, 1FIN, 1FQ1, 1FVT, 1FVV, 1G5S, 1GIH, 1GII, 1GIJ, 1GY3, 1GZ8, 1H00, 1H01, 1H07, 1H08). To identify the reference structure, use the Reference Structure Inputs parameters. Only one reference structure can be used per floe, and a reference structure can be identified using a dataset, pdb and mtz files, or a PDB code. For this example we use the CDK2 PDB code from above (5K4J) as the reference DU:
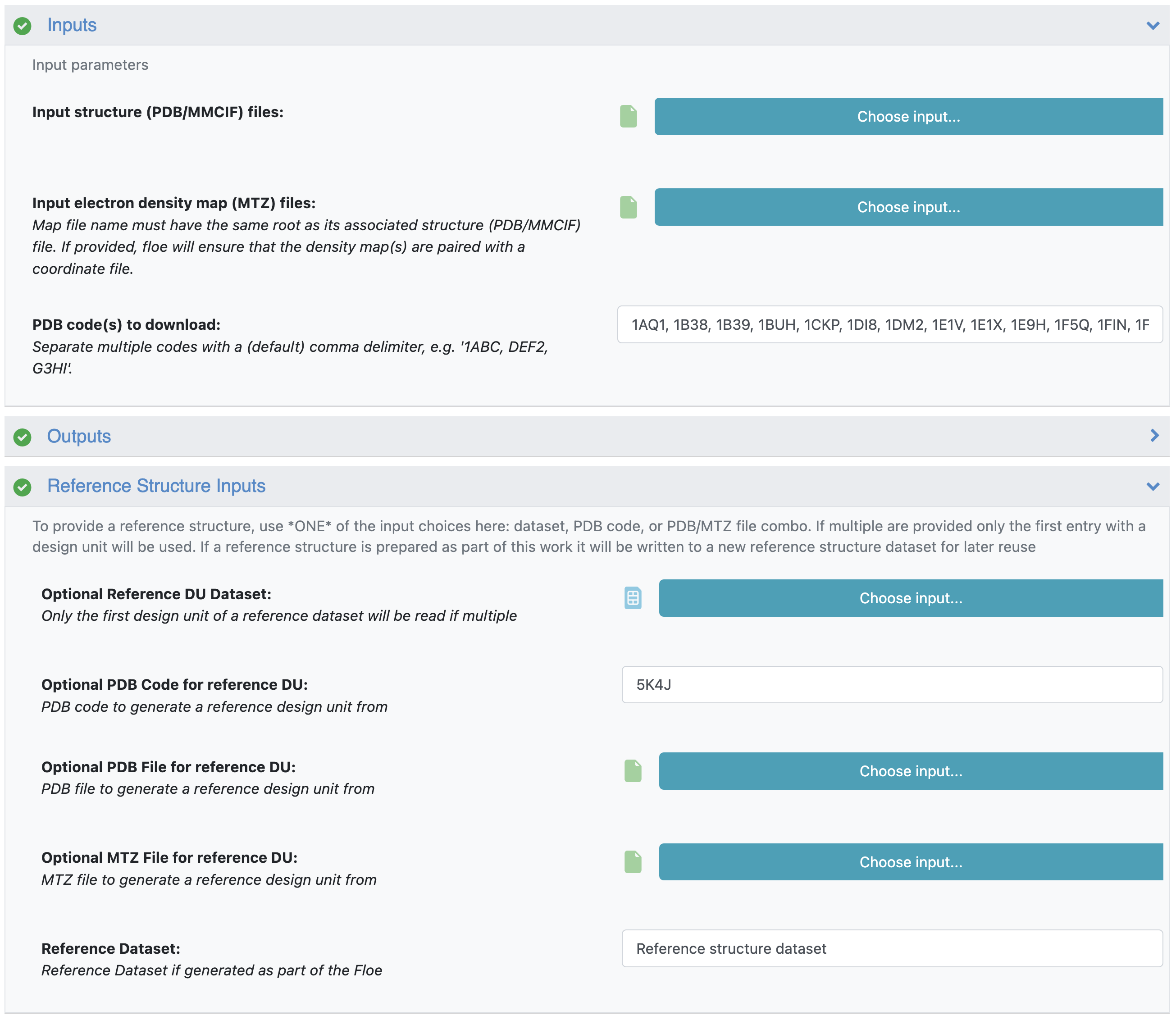
CDK2 subset prep with reference DU
Reviewing the Floe Reports
This results in a dataset with 32 design units, and a dataset with three failed records. The failures are due to reference structure mismatches or inconsistencies in the structure, and are filtered out with default settings. A dataset containing the reference structure is also created.
The 32 prepared design units from 24 PDB files are a result of multiple biological units generated from the crystal structure asymmetric unit, and certain experiments with alternate locations (configurations) near or in the binding site of interest. A floe report is also generated detailing issues with the prepared design units that may need further inspection.

Floe report continues with more potential issues after structure preparation
Analyzing the Results
First, we are going to mark both the reference DU dataset and the dataset containing the prepared DUs as active.
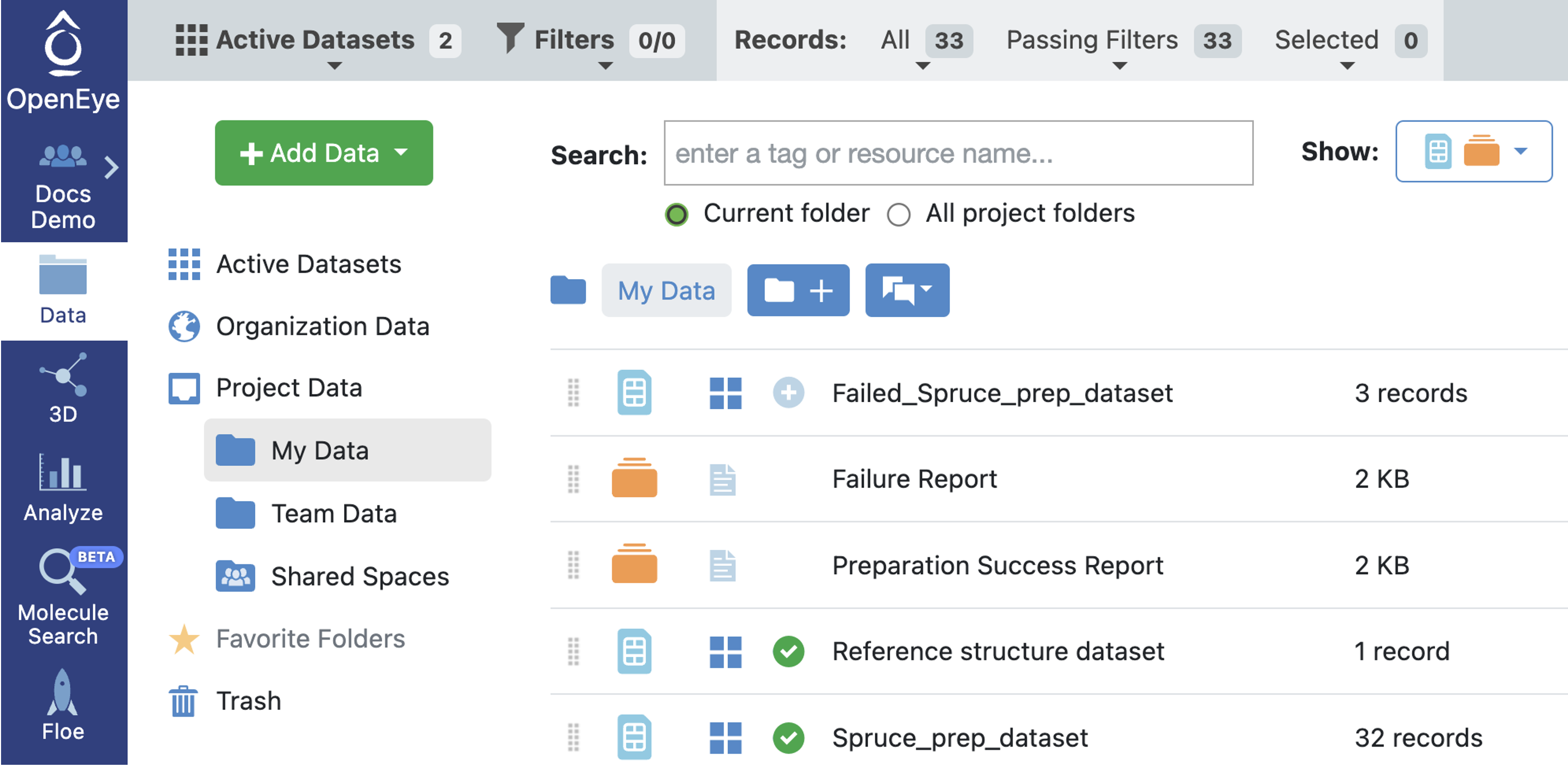
Activate datasets
When analyzing a dataset containing a larger number of records, it can be helpful to view them in the analyze page. This makes it easy to explore and filter on a property such as the Iridium Classification. In this case, we have filtered out NA (Not Applicable, no Iridium Score could be calculated) and NT (Not Trustworthy) structures. Furthermore, the remaining structures have been sorted on the classification. This filtering can also be done in the 3D viewer, however, here we use the analyze page because we can also sort the records based on the Iridium classification and we can inspect the various depiction images, giving us a high level overview of the dataset before switching to the 3D view.
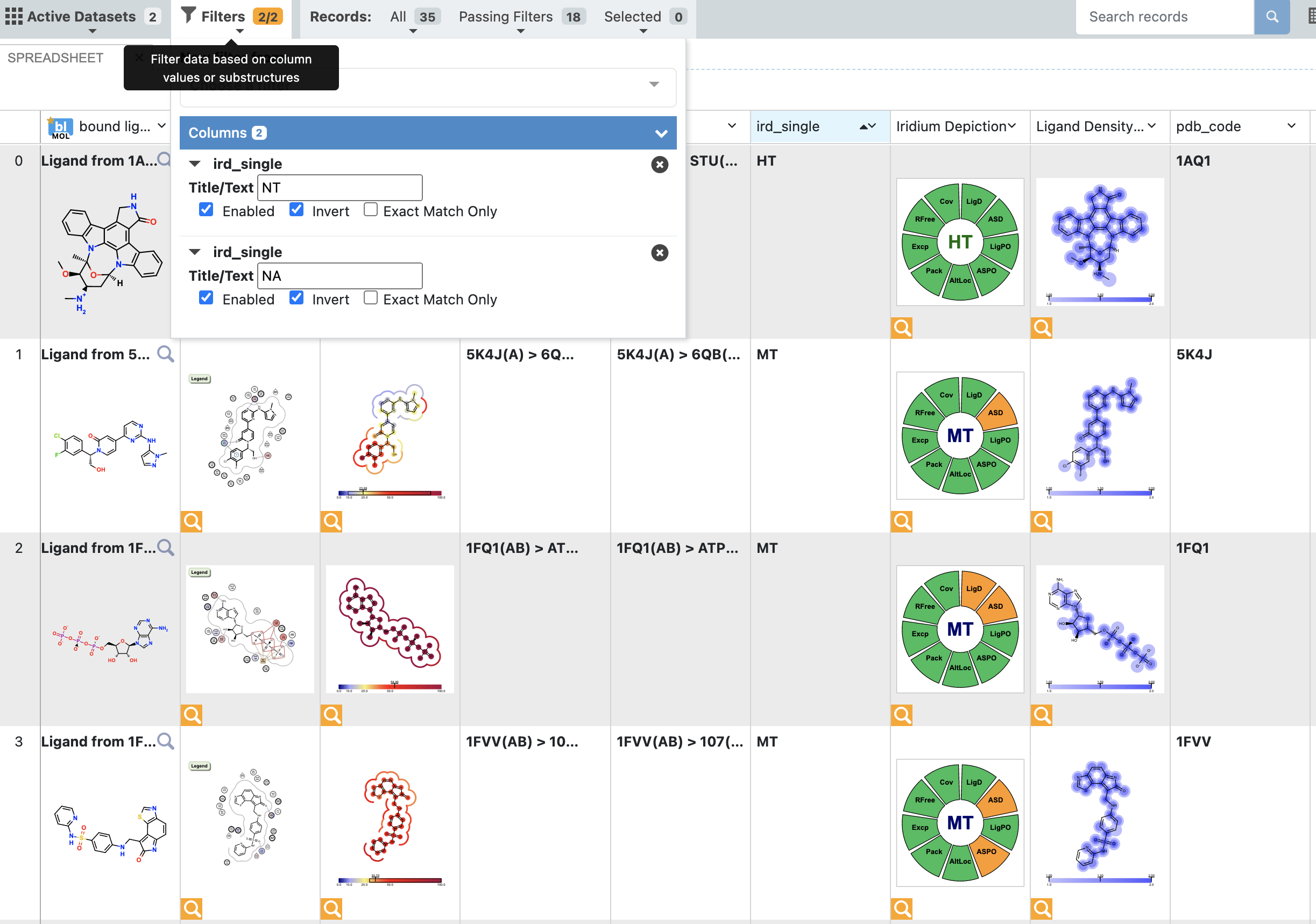
Filter dataset
Once filtering has been applied, the relevant structures can be viewed on the 3D page. Since a reference structure was used, all the structures are superposed, allowing for easy comparison and exploration of differences in ligand binding poses or binding site conformations. In the figure below, the reference structure dataset is shown, with protein-ligand interactions indicated.
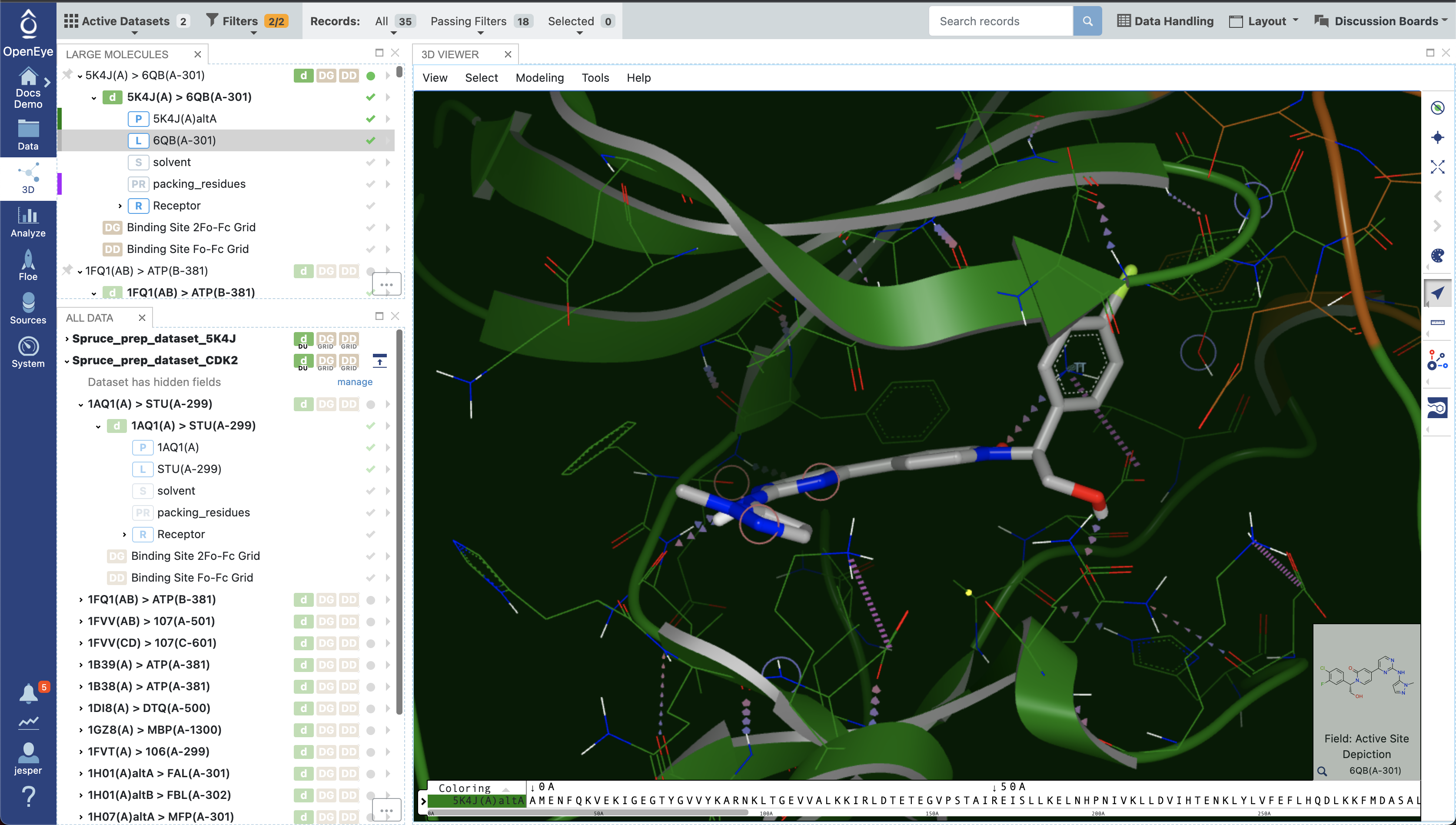
Design Unit in 3D viewer
Data stored on the DU record makes it possible to inspect the experimental electron density maps with just a few mouse clicks.
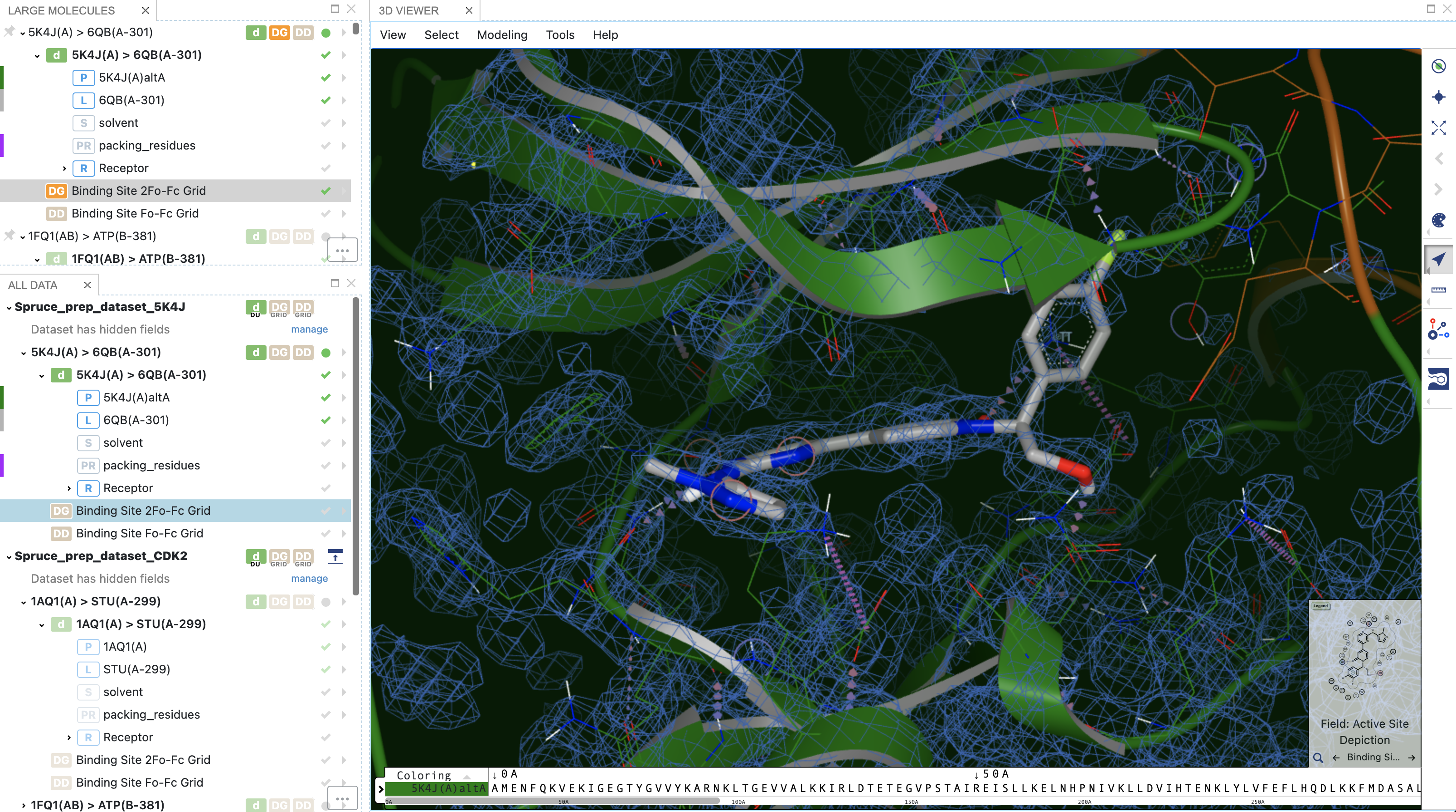
2Fo-Fc electron density grids shown with the structure
Multiple structures may be displayed at the same time, using different color schemes for the bounds ligands. Inspecting differences in binding poses, particularly specific binding interactions such as hydrogen bonds, salt-bridges, and pi-type interactions, can be helpful in explaining experimental data, for example differences in activities, or differences in specificity of a compound between different targets.

Design units in a dataset with a reference DU are superposed. One of the bound ligands is shown with a different color scheme.
Depending on the subsequent modeling task, a single or multiple design units can be selected and saved as a new and curated dataset. In this case, we could save only the structures with an HT (Highly Trustworthy) Iridium classification. It is important to note that many MT structures are also relevant for use in modeling tasks. The MT classification indicates that a structure should be inspected closely prior to being used in computations, in particular the ligand binding site(s), to avoid unexpected results.
Calculation of the Iridium Classification requires electron density maps to be supplied in MTZ format. If these maps are not available, the Iridium classification cannot be calculated, and Classification is NA. Furthermore, Iridium cannot be calculated for apo structures, which are also marked NA.
Next Steps
Design Units prepared by Spruce can be used for a variety of modeling tasks. One integral part of the DU preparation process is the generation of a receptor binding site (docking grid), which allows the DU to be used for ligand docking or posing tasks.