Introduction
Full List of Crystal Math Floes
In this tutorial, polymorphs of benzoic acid will be predicted using the crystal structure prediction (CSP) Floes in the Crystal Math Floes package. There are total 18 Floes available in this package. They can be found in the Product-based / Crystal Structure Prediction category path as shown below:
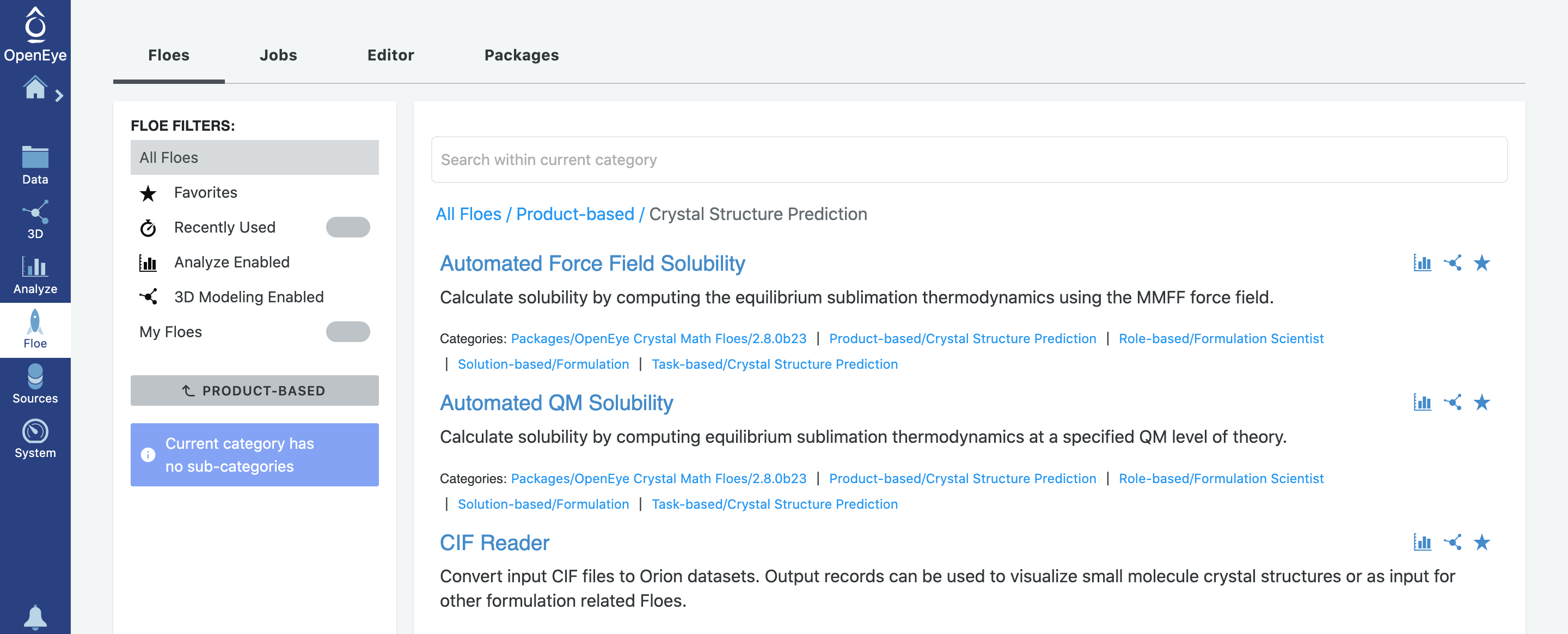
Information about this Tutorial
There are four Main Floes in the CSP Protocol used to predict crystal structures and rank them based on QM enthalpies at 0 K. Then, a description of Additional Optional Floes is provided, which include Floes to correct the energy landscape to finite temperature and to compare predicted crystal structures to experimental crystal structures or X-ray powder diffraction patterns.
Running all the Main Floes in the CSP Protocol in this tutorial will cost approximately $1,300 on Orion with some variation based on AWS instance availability. Reducing the number of crystal structures predicted or optimized at any stage will reduce the total cost of the tutorial. Follow these Cost Saving Options at each stage to reduce the total cost to approximately $30. These options are provided to make the tutorial cheaper, but would not be recommended for a prospective prediction. In addition, the output from all Floes is included in the tutorial data described below.
Keep in mind that benzoic acid is a very small and rigid molecule. The costs of this tutorial are much lower than would be expected for a drug like molecule. When starting predictions with a new molecule, the Cost Estimate Floe can help with understanding how much a full prediction will cost.
Tutorial Data on Orion
For every Floe described in this tutorial the input and output is available in Organization Data on Orion. From the Data page, select “Organization Data” on the left. Then navigate through the folders “OpenEye Data” –> “Tutorial Data” –> “Crystal Math Floes.” The image below shows the folders for this tutorial:
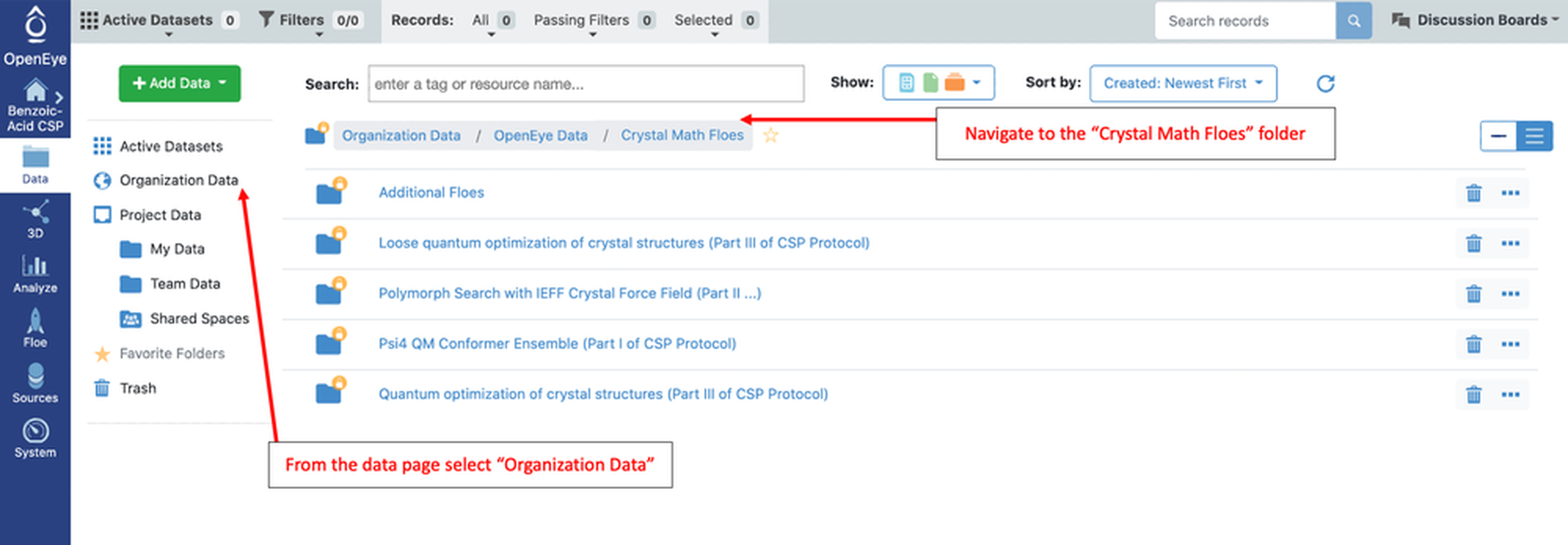
Each sub-folder is named after a Crystal Math Floe. The output from the Floe is stored in the corresponding folder. In this tutorial, the input for most Floes will be the output from a previous step. When that is not the case, the input dataset is also included in that Floe’s folder (as indicated below). In the top level folder there is output for the four Main Floes in the CSP Protocol:
Psi4 QM Conformer Ensemble (Part I of CSP Protocol) (includes benzoic-acid input dataset from SMILES file)
Polymorph Search with IEFF Crystal Force Field (Part II of CSP Protocol: Generation and Filtering)
Loose Quantum Optimization of Crystal Structures (Part IIIA of CSP Protocol)
Quantum Optimization of Crystal Structures (Part IIIB of CSP Protocol)
Then, in the “Additional Floes” folder there are more sub-folders for some of the Additional Optional Floes:
CIF Reader (includes experimental CIF file)
Filtering of Crystal Structures based on Powder Diffraction Pattern (includes XYE file)
QM crystal entropy with a cluster expansion method (Part IV of CSP Protocol)
For this tutorial, the experimental benzoic acid CIF file 7210454.cif was taken from the Crystallography Open Database. The CIF file was then parsed with the CIF Reader Floe to view and use the crystal structure on Orion. A theoretical X-ray powder diffraction pattern was created with OpenEye tools and can be used as an example in the Filtering of Crystal Structures based on Powder Diffraction Pattern Floe.
The only required input for crystal structure prediction is a SMILES
of your molecule. So an input dataset can be created with the Orion Sketcher.
From the data tab, in the top left click on “+Add Data” then choose
sketch. In this box you can also paste the smiles, such as
c1ccc(cc1)C(=O)O to be used in this tutorial.