Theory
Having reliable and fast crystal structure prediction (CSP) and solubility prediction of drugs is of great importance for the drug industry. This OpenEye Orion Workfloe Package is a collection of tools for making such predictions.
Crystal structure predictions
The protocol for finding the most stable crystal structure is to first apply Force Field – faster but less accurate method – to sample the crystal structure space. To achieve precise predictions, several crystal structures, which have lowest lattice energies according to Force Field analysis, need to be optimized and re-ranked in energy with more accurate approaches (also more expensive), such as the densitiy functional theory (DFT) method. The structure corresponding to lowest lattice energy after re-ranking would be the theoretical prediction of most stable crystal structure.
Random packing
The sampling in CSP consists of two stages. At first stage the three dimensional geometry of conformation of drug-molecule is being generated from the SMILES string. At the second stage a crystal spacegroup symmetry is chosen, and each generated conformation is packed into the crystal unit cell, according to the selected crystal symmetry. The former is achieved with openeye toolkit Omega, while the latter tool is released with this Orion package.
Packing can be achieved with and without some knowledge about the unit cell parameters. In practice, sometimes such knowledge is present from powder diffraction. In such case the unit cell parameters are assumed to be known and we allow such functionality in the PackPrep Cube. The result of packing is the desired number of randomly generated crystal structures which all pass a robust bump check which ensures no clashes of atoms, keeping the periodic boundary conditions in mind.
Force field
Traditional Force Fields, such as MMFF94 and AMBER are known to poorly describe intermolecular energies. OpenEye has developed IEFF Force Field, which is an extension of MMFF94 with intermolecular Coulomb and Van der Waals energies. Former is based on (up to quadrupole) multipoles, which can be obtained from quantum wavefunction of appropriate level of theory. The latter energy uses parameterised form with not too many (3) parameters per atom. These parameters have been fitted to CCSD dimer energy-distance curves.
IEFF Lattice is a specific extension of IEFF Force Field adapted for crystal lattice energy calculations. It is included in this Orion package in the form of the ‘IEFF Lattice’ Cube which allows for lattice energy calculation with intermolecular energy computed by IEFF, and also including long-range summation of Coulomb interactions, via technique known as Ewald summation.
Quantum Methods
Below we review three types of quantum methods for crystal structure energy evaluation. First one is based on semi-empirical quantum approach. Second one is a powerful new method developed at OpenEye based on splitting the crystal energy into sum of independent dimer energies. Third approach is the traditional Plane-Wave DFT with periodic boundary conditions. In the current release we only provide tools for the second approach, for DFT optimization with parallel dimers computations at each step of optimization.
Semi-empirical
Semi-empirical quantum approaches have the speed advantage, especially when compared with ab-initio plane-wave DFT. However this benefit comes at a price: the accuracy of such methods is not significantly better than that of Force Field.
We use open source code Density Functional based Tight Binding (DFTB+). For appropriate lattice energy calculation, the atom-pariwise D3 dispersion correction in the Becke-Johnson damping variant needs to be switched on. This software allows for taking into account periodic boundary conditions. In Ref. [Brandenburg-2014] this package was used to successfully predict lattice energies for 21 crystal structures.
Cluster of Dimers
In this approach we utilize the expansion of lattice energy into many-body interaction energy terms. Unless monomers in the crystal are extrelemely close to each other, the dimer approximation is usually sufficient to obtain robust estimates of crystal energies and sublimation thermodynamics. The central monomer is chosen and cluster of dimers is built, each sharing the central monomer and with distance between monomers in each dimer within some radius (typically 6-8A).
The power of this method is that energies of different dimers can be computed in parallel, thus taking full advantage of cloud computing with this Orion package. With a given theory level of DFT, the lattice energy computed using parallel dimer energies calculations can be computed with a speedup of ~50-100. This is cruicial for reducing the CSP timing from several weeks to ~ 1 day.
In addition to this speedup, by adjusting the level of DFT method and the size of basis set we are aiming to obtain full crystal structure and solubility predictions overnight.
The quantum package of our choice for dimer energies is Psi4 – an open source quantum chemistry package for DFT, MP2, SAPT, and Coupled-Cluster calculations. We include into this Orion package a Psi4 Cube for DFT calculations, and the QM multipoles Cube, that uses Psi4 to compute electric multipoles, in order to enable the user with subsequent IEFF Force Field application.
Plane-Wave DFT
This is a well established approach for describing infinite periodic systems. It uses Bloch’s theorem, k-point sampling, and pseudopotential approximation for describing core electrons. Open-source package Quantum Espresso is a well-known package for Plane-Wave DFT calculations.
While this is the most accurate method fo lattice energy computations, it is extremeley slow, scales poorly with the size of the system, and is not breakable into parallel computations.
Intrinsic Solubility
Intrinsic aqueous solubility is a measure of equilibrium between solid and liquid states of a molecule. A well established approach for solubility calculation is to describe solid to liquid transition of the molecule via a two-step transition of solid to gas at normal conditions, followed by gas to liquid transition. This is schematically shown in the figure below. Consequently the thermodynamic quantities equal to the sum of sublimation and hydration terms.
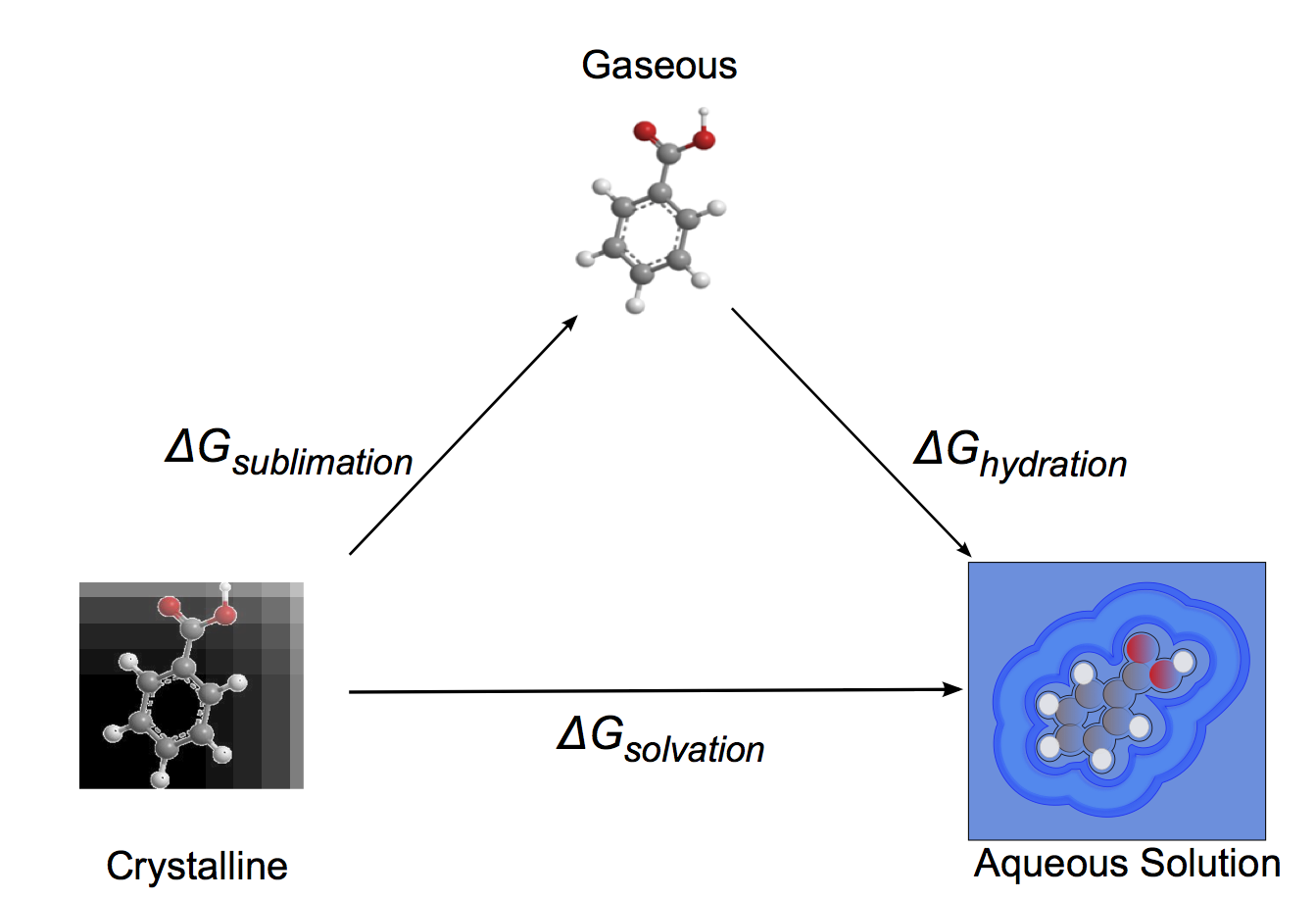
Thermodynamic cycle for predicting intrinsic solubility
Free energy of hydration for drug-like molecules is well described by the OpenEye toolkit Zap. This package includes new set of tools developed at OpenEye for sublimation free energy calculations, which equals
Enthalpy is well approximated by sum of the lattice energy and the \(-2RT\) term [Gavezzotti-1997], [Otero-de-la-Roza-2012]
where \(R = 8.314 \,\text{J}/(\text{mol} \,\text{K})\) is the gas constant and \(T\) is the temperature of the system in Kelvin. Entropy of sublimation is the change in entropy per monomer during the sublimation process. It equals:
In the equation above \(S^0_{\text{vacuum}}\) is the configurational entropy from Szybki toolkit, \(S^{\text{unit cell}}\) is the crystal phonon entropy of unit cell, and \(Z\) is the number of monomers in the unit cell. In Refs. [McDonagh-2016], [Cervinka-2016] thermodynamic sublimation properties calculated from physics methods are shown to be in good agreement with experimental data.
Finally, solubility in terms of sublimation and hydration free energies equals to [Palmer-2012]
where \(p_0\) is the atmospheric pressure.