Additional Optional Floes
For each of these Floes, if the cost is not described, assume it is trivial (well under $1 in most cases). For example, the cost of performing 200 RMSD20 calculations for this tutorial was approximately $0.05.
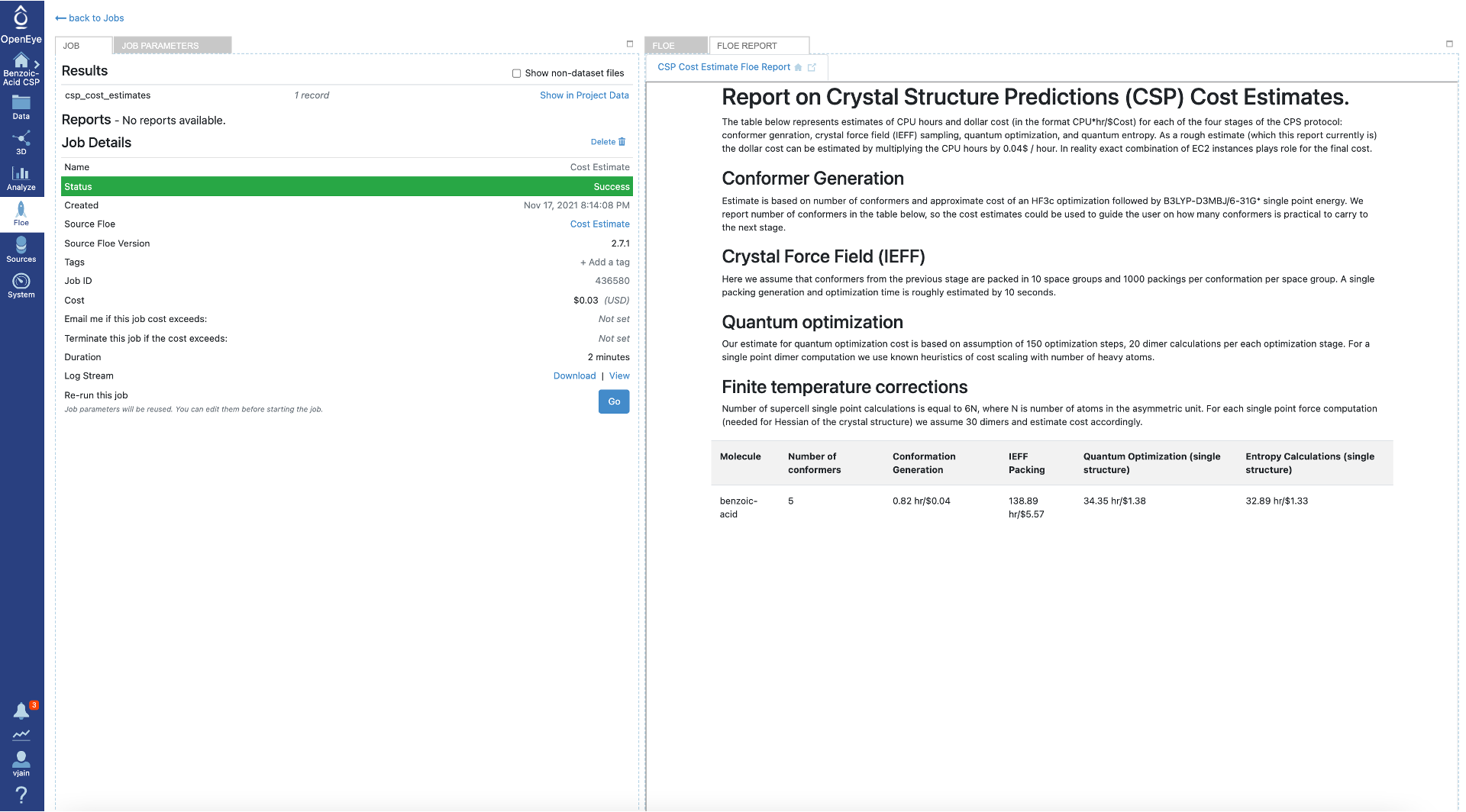
CIF Reader
The CIF Reader Floe converts a CIF file into an Orion dataset, which is necessary for viewing crystal structures in the Orion 3D viewer or using that crystal structure as input to any Floe. For example, parsing an experimental CIF file allows it to be used as the reference in the Crystal RMSD Floe. Upload all CIF files to be parsed to Orion. Multiple CIF files can be used as input, the resulting structures are saved to a single dataset.
Note
At this time, this Floe only supports CIF files with a single structure. CIF files with multiple structure will need to be separated before running the Floe.
The image below shows the interface of CIF Reader Floe. A CIF file for benzoic acid is available in the Crystal Math Floes tutorial data (for details about Tutorial Data on Orion).
The crystal structures can be seen in the 3D viewer by selecting the primary Molecule and either the “Crystal (small) Cluster” or “Crystal Cluster” fields. Example output from this Floe is also available in the tutorial data.
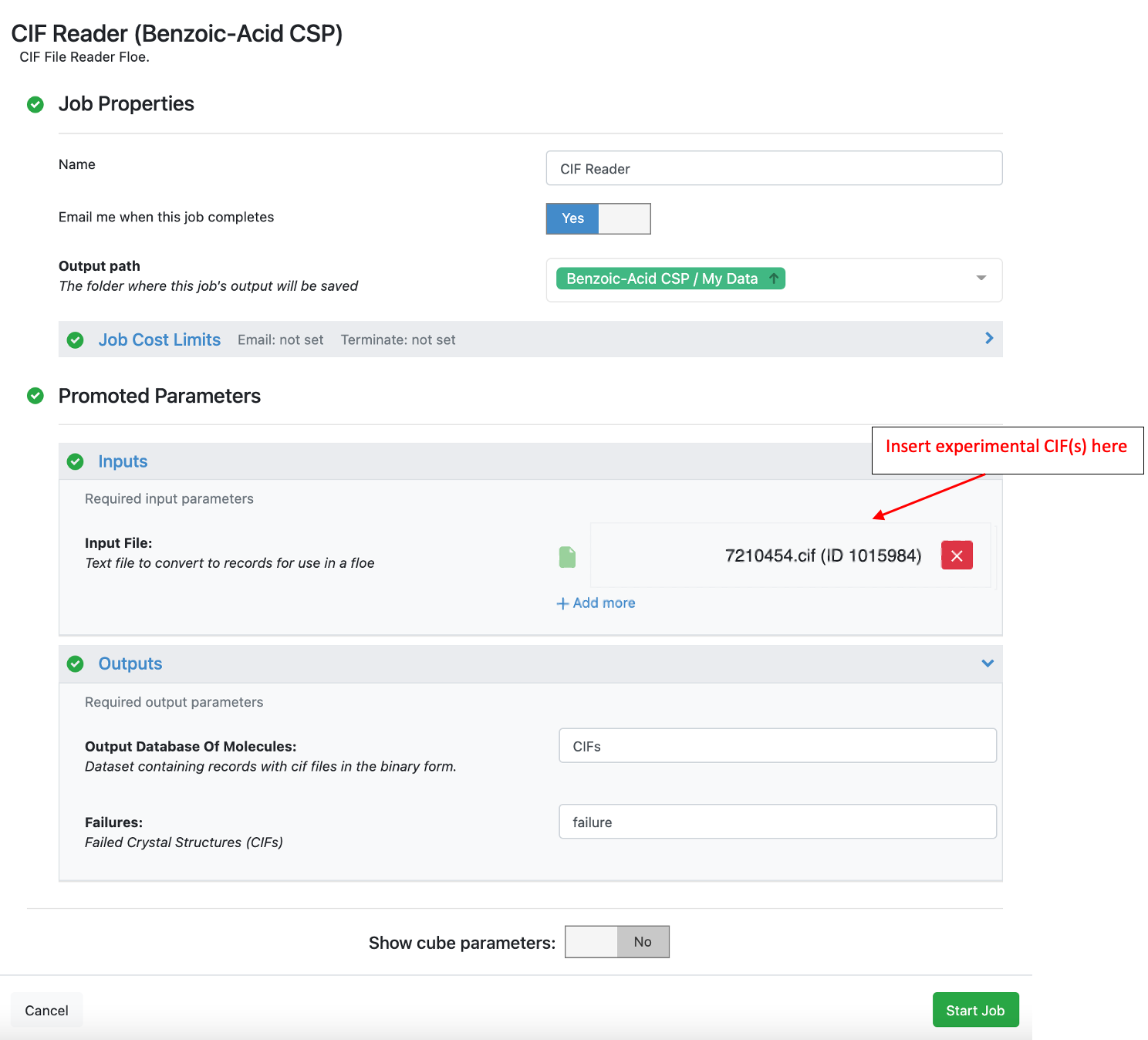
Crystal RMSD Floe
The Crystal RMSD Floe compares a reference crystal structure(s) to several predicted (Fit) crystal structures. Both reference and fit structures should be CIF strings on the records. Any predicted structures from these CSP Floes or those generated with the CIF Reader will meet this requirement. By default, this Floe calculates RMSD20 which is the root mean squared deviation for the overlay of matching clusters of 20 molecules from the reference and predicted crystal structures. This calculation can be performed after Part II or Part III (A or B) of CSP protocol.
In this tutorial, the RMSD20 was calculated between the experimental and predicted structures of benzoic acid. Both input datasets can be found in the tutorial data the “CIFs” dataset comes from the “Additional Floes/CIF Reader” folder and the “qm_optimized” dataset is in the “Quantum optimization of crystal structures (Part IIIB of CSP Protocol)” folder.
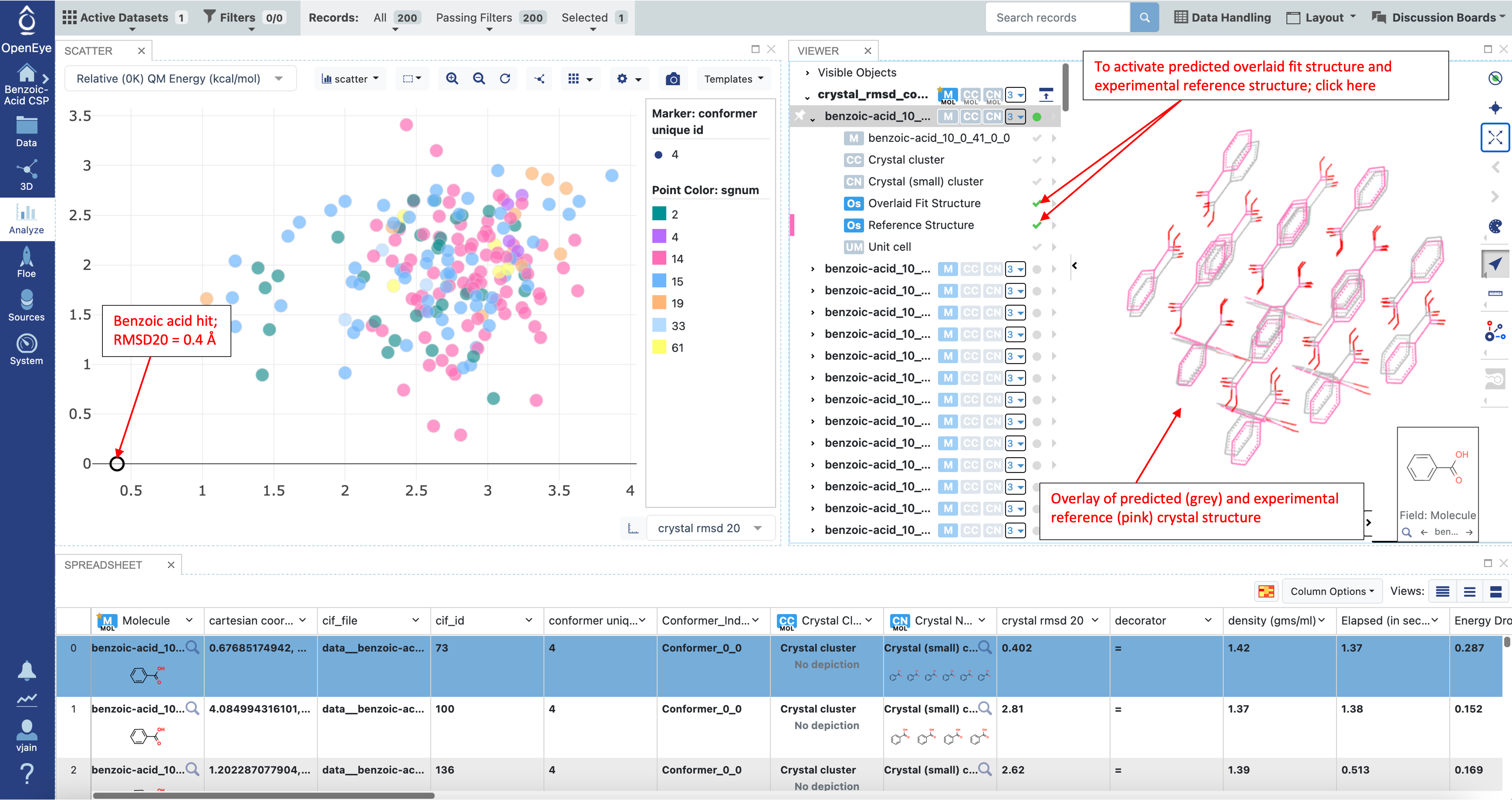
The output dataset, crystal_rmsd_compared contains the crystal RMSD20 between experimental and predicted (Fit) structures. Generally, any RMSD20 less than 0.5 angstroms is considered identical to experiment. Therefore, the structure with an RMSD20 of 0.4 A shown below is considered a hit for benzoic acid. To visualize overlays of predicted and experimental structures select the “Overlaid Fit Structure” and “Reference Structure” fields on the output record in any 3D viewer on Orion.
Example output from this Floe can also be found in the tutorial data in the “Additional Floes/Crystal RMSD Floe” folder.
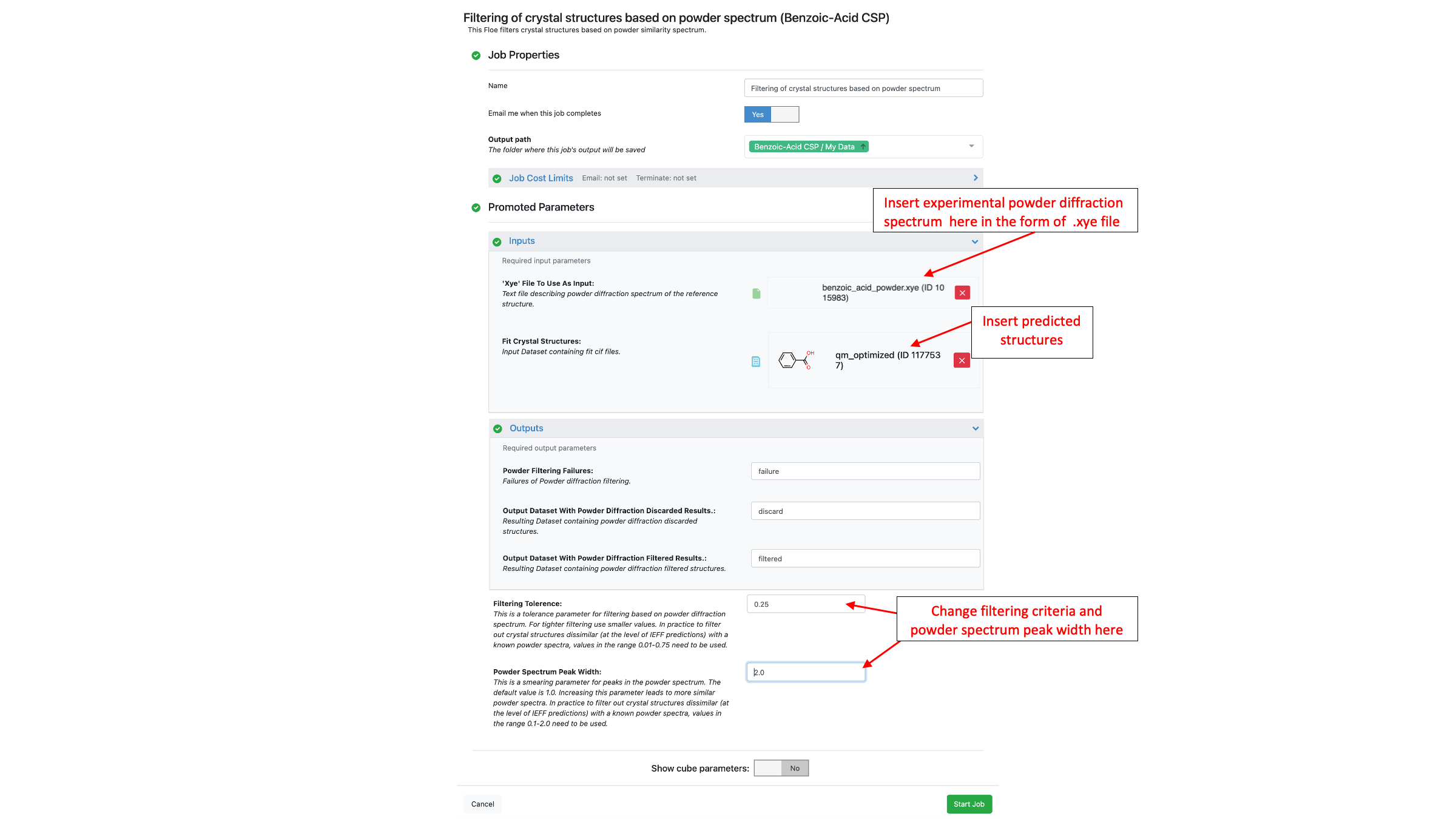
Filtering of Crystal Structures based on Powder Diffraction Pattern
The Filtering of Crystal Structures Based on Powder Diffraction Pattern Floe compares predicted crystal structures to a powder X-Ray diffraction pattern. For each predicted structure, a theoretical diffraction pattern is calculated. Then, the similarity between the predicted and experimental diffraction patterns is calculated as a correlation. The final score for each predicted structure is reported as “1-similarity” meaning the closer a score is to 0.0 the more similar the structure is to the experimental PXRD. The “Filtering Tolerance” parameter determines how similar a structure needs to be to be considered a match. Any score less than this value is sent to the “Output Dataset With Powder Diffraction Filtered Results” and any score above this threshold is sent to the “Output Dataset With Powder Diffraction Discarded Results”. If a powder diffraction pattern cannot be calculated for a crystal structure then it will be sent to the “Powder Filtering Failures” output (which is rare). The XYE file for benzoic acid is available in the tutorial data in the “Additional Floes/Filtering of crystal structures based on powder diffraction pattern” folder. For the Fit Crystal Structures input, the “qm_optimized” dataset from Part IIIB of this tutorial is used, which is also available in the “Quantum optimization of crystal structures (Part IIIB of CSP Protocol)” folder.
Note
This Floe currently only supports XYE input with 2-Theta starting at 5.00 and step sizes of 0.02. Future releases will include support for more file formats and will be able to process the input of different step sizes automatically.
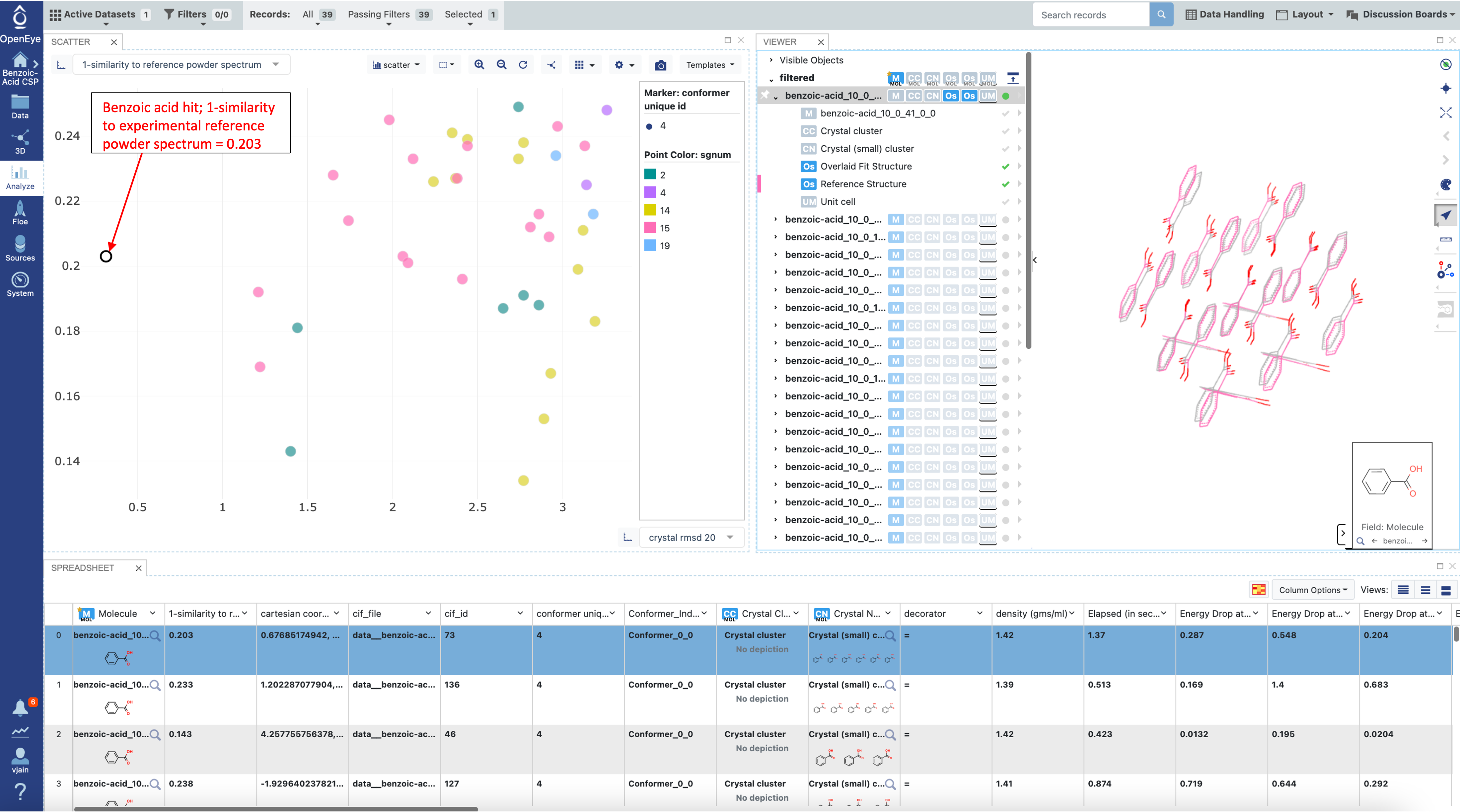
Of the initial 200 crystal structures 39 are considered a match (have a score of less than 0.25 when comparing the diffraction pattern) and 161 are discarded because their score is above the threshold. In the image below, RMSD20 is compared to the PXRD similarity for these same predicted structures and corresponding experimental references.
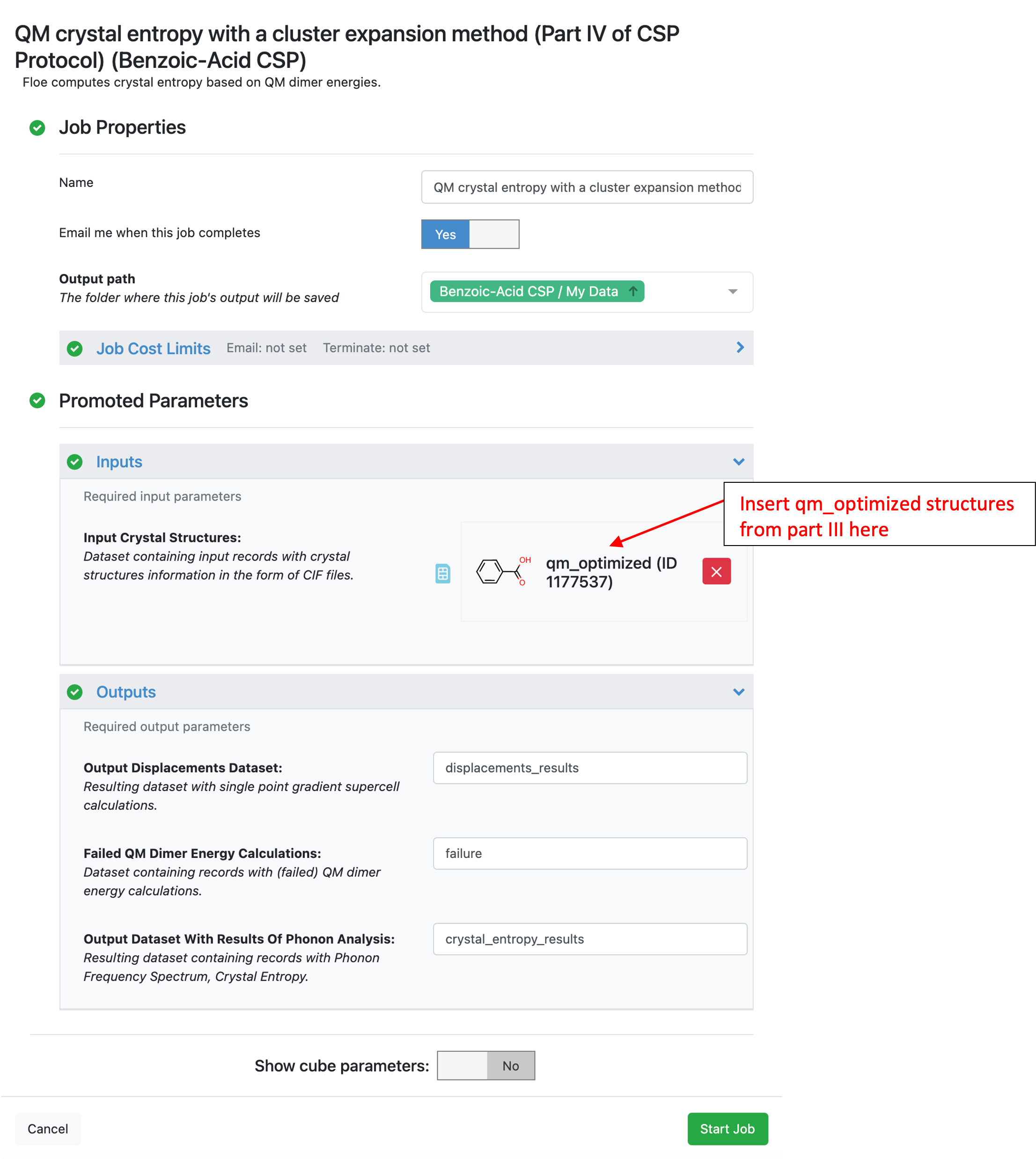
QM crystal entropy with a cluster expansion method (Part IV of CSP Protocol)
The QM Crystal Entropy with a Cluster Expansion Method (Part IV of CSP Protocol) Floe provides a method to calculate CSP energy landscapes at finite temperature by calculating the phonon free energy contribution. The input to this Floe should be the tight optimized structures from Part IIIB of the CSP protocol.
The 13 lowest energy structures of benzoic acid (corresponding to a 1 kcal/mol energy window) were used for this tutorial. The input structures came from the “qm_optimized” data set in the “Quantum optimization of crystal structures (Part IIIB of CSP Protocol)” folder. Example output from this Floe is available in the tutorial data in the “Additional Floes/QM crystal entropy with a cluster expansion method (Part IV…)” folder.
The cost to calculate the HF-3c entropy for these 13 structures is about $38 or approximately $3 per structure. The cost of Entropy calculations can be quite significant (up to $100/calculation) as they require many QM gradient calculations. The Cost Estimate Floe includes an estimate of the cost per structure.
Free energies are computed at 300K and reported in the “Relative Room Temperature Free Energy (kcal/mol)” field in the results.
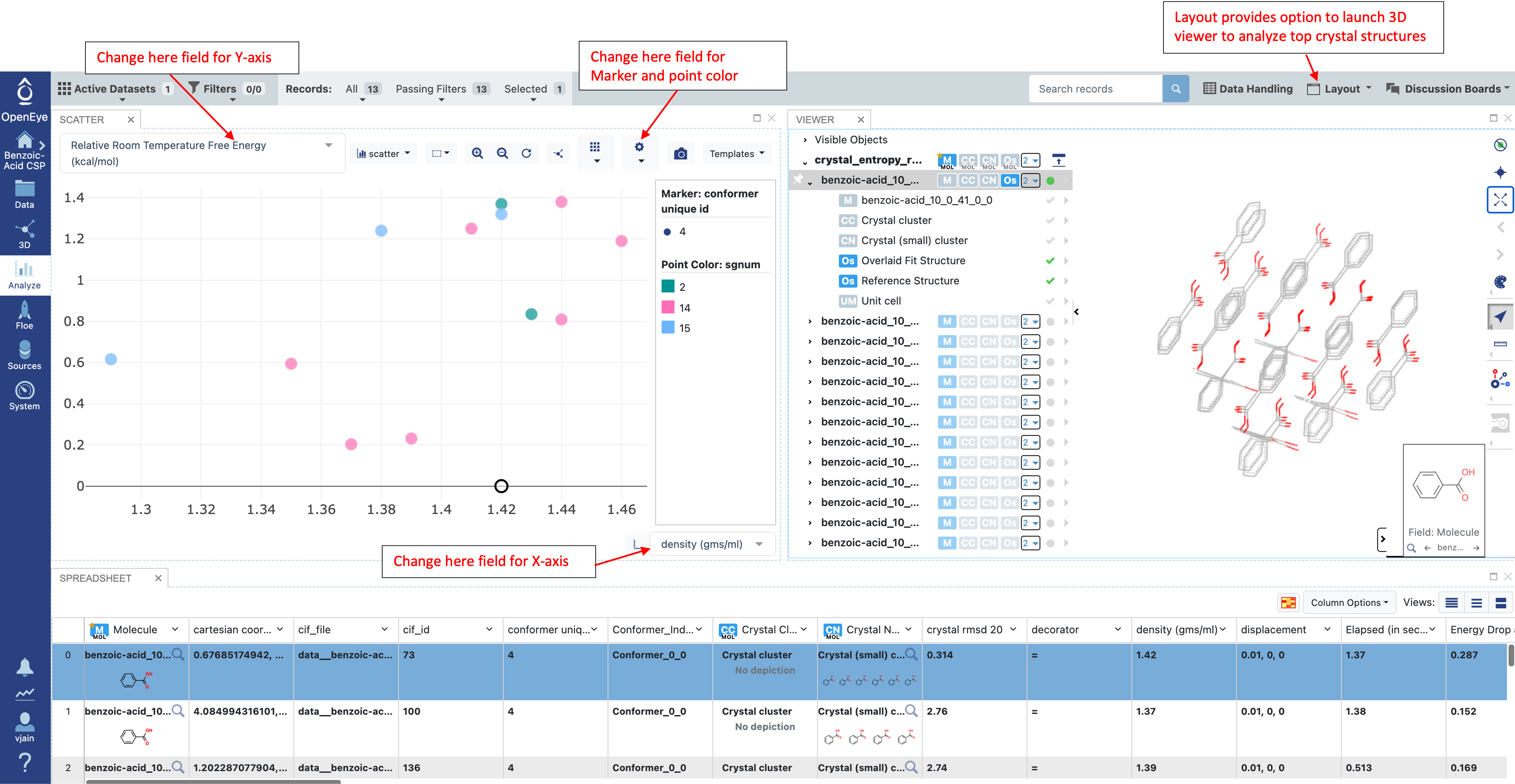
An energy window from 1-2 kcal/mol is used to filter results from Part IIIB. Due to the cost of QM entropy calculations, computing the finite temperature correction for all predicted structures is usually not practical. However, the finite temperature contribution can be computed at a force field level of theory for all structures. For a prospective prediction, all structures from Part IIIB would be corrected using a force field entropy calculation. Structures in the low energy range or whose rank changed significantly with the force field finite temperature correction are included in the QM entropy calculation.
Force Field Optimization of Crystal Structures in the Dimer Expansion Search
The Force Field Optimization of Crystal Structures in the Dimer Expansion Approach Floe is used to optimized crystal structures with the MMFF force field. In order to compute force field based crystal entropies, the structures must be in a local minima for that level of theory, so an optimization needs to be performed first. The input to this Floe can be any crystal structure dataset on Orion.
The cost of optimizing structures with force fields is very cheap compared to QM, typically under $0.50 per structure.
Force Field Crystal Entropy with a Cluster Expansion Method
The Force Field Crystal Entropy with a Cluster Expansion Method Floe provides the option to correct CSP energy landscapes at finite temperature with the incorporation of MMFF force field-based crystal entropies. However, input should be the force field optimized crystal structures from The Force Field Optimization of Crystal Structures in the Dimer Expansion Approach Floe. This Floe allows Crystal entropies for finite temperature correction to be computed at a very low cost. For prospective calculations force field entropies are always computed before QM entropies and help guide the filtering process.
The cost of calculating entropy with force fields is very cheap compared to QM, typically less than $0.50 per structure.
Automated QM Solubility
The Automated QM Solubility Floe performs all three steps to compute the solubility of a crystal:
optimizes input crystal structures,
calculates entropy in order to determine the finite temperature sublimation free energy, and
uses Zap to compute the hydration free energy.
The result is a fully calculated thermodynamic equilibrium solubility. Levels of theory are B3LYP-D3/6-31G* for sublimation enthalpy with HF-3C for geometry optimization and phonon free energy.
The cost of this Floe will be significant because it requires performing a QM optimization, single point energy, and entropy calculation. For benzoic acid it would run about $7 per structure, but that cost will increase based on the number of atoms in the molecule. To approximate this cost, run the Cost Estimate and combine the cost of the Quantum Optimization of Crystal Structures (Part IIIB of CSP Protocol) and QM crystal entropy with a cluster expansion method (Part IV of CSP Protocol) Floes.
Automated Force Field Solubility
The Automated Force Field Solubility Floe performs the same three calculations described above, but with a force field level of theory (MMFF) for sublimation enthalpy and phonon free energy.
Similar to other force field Floes, the cost of this combined Floe will be much cheaper than running QM calculations, likely under $1 per structure.
Solubility
The Solubility from Crystal Entropy and Enthalpy Floe computes thermodynamic equilibrium solubility. The input is assumed to contain
optimized crystal structures with sublimation enthalpy (output from CSP Part IIIB),
phonon free energy (from one of the entropy Floes) and
hydration free energy.
If these values have not been computed, then use the Automated QM Solubility or Automated Force Field Solubility Floes where all of these pieces are computed automatically.
Polymorph Filtering based on IEFF Energies (Part II’ of CSP Protocol: Filtering)
The Polymorph Filtering Based on IEFF Energies (Part II’ of CSP Protocol: Filtering) Floe provides a way to re-filter IEFF top crystal structures from a collection created by Part II of CSP protocol. Multiple collections from Part II can also be used as input to filter all of their results in to one set of top structures. If the multi-stage protocol is used where the Polymorph Search with IEFF Crystal Force Field (Part II of CSP Protocol: Generation and Filtering) Floe is run multiple times, then all of the resulting collections should be combined and re-filtered before moving to Part IIIA of the protocol.
There is an example collection, “IEFF Crystal Packings Collection,” in the “Polymorph Search with IEFF Crystal Force Field (Part II …)” folder of the tutorial data which can be used as input for this Floe.
The cost of this Floe will range dramatically depending on the size of the collection, chosen energy window, and number of structures to deduplicate. The cost will always be a small fraction of the original IEFF predictions. Re-filtering the provided collection (50,000 structures) with all default parameters costs about $1.50. For an alternative example, a collection with around 200 million crystal structures will cost around $100.
Psi4 Combined Tautomer and Torsion Sampling Conformer Floe
The Psi4 Combined Tautomer and Torsion Sampling Conformer Floe Floe contains 3 parts:
All reasonable tautomers of the input molecules are enumerated.
Fragments are generated around each single-bond in each tautomer. These fragments are created to mimic the energetics of the torsion around the single-bond in the original tautomer. Torsion scanning is performed with the OETorsionScan function from the OESzybki Toolkit at a specified resolution (in degrees), no less than 10 should be used for this Floe. This function includes a force field minimization of all internal degrees of freedom constraining the rotatable torsion in each fragment. Then, a QM optimization is performed with the torsion constrained while all other degrees of freedom are relaxed. Lastly, the energy landscape of the torsion scan is used to determine custom sampling rules for that torsion.
Conformers are generated with the specified RMSD threshold using the custom sampling rules determined by the torsion scans. These conformers are optimized with all torsions fixed (HF-3c) and then a single point energy is performed at a higher level of theory (B3LYP-D3MBJ/6-31G*). Conformers at the Omega level (no QM optimization) are also generated at lower thresholds to determine if a multi-stage or single stage approach should be used for the prediction.
Finally, a report is generated summarizing the results from all of these steps. This report includes an approximation of how many conformers will be in final ensembles at higher resolutions. Generally for CSP, conformers are desired at a 0.25 A threshold. However if the number of conformers at that level is too large, then the multi-level approach might be more desirable.
There are five dataset outputs (and the Floe Report) for this Floe:
“Torsion Rules Output”: All enumerated tautomers with the custom torsion sampling rules saved on the records. This output can be used as input to the Psi4 QM Conformer Ensemble (Part I of CSP Protocol) Floe to use the custom sampling rules in conformer generation.
“Fragment Output”: All fragments for all tautomers. This output is a multi-conformer molecule with one conformer for each angle sampled during the torsion scan.
“Intermediate Optimization Output”: One record for every optimized conformer. These are saved so that all optimized conformers are in one dataset.
“Psi4 Conformer Ensemble Output”: Optimized conformers with calculated single point energies which fall within the specified energy cutoff.
“Failure Output”: Failures for any step in the Floe. Most of these will be very high energy conformers which fail to optimized from either the conformer ensemble or the torsion scans.
The cost for this Floe will be of a similar magnitude to other Conformer Ensemble Floes. With the additional cost of running torsion scans on fragments. For example, it costs about $0.75 to run with benzoic acid as an input with all default parameters.
Water Sampling Floe
The Water Sampling Floe Floe takes as input a list of conformers and samples water positions around them to generates mono-hydrate dimers. The generated output can be sent to the Polymorph Search with IEFF Crystal Force Field (Part II of CSP Protocol: Generation and Filtering) Floe to predict crystal structures of mono-hydrates.
The bulk of the cost for this Floe is calculating QM multipoles for each conformer and should be relatively cheap in general. For example, using the five benzoic acid conformers from the Part I tutorial data as input with all other parameters set to default costs around $0.25.
Cost Estimate
The Cost Estimation for Crystal Structure Prediction Floe performs a cost estimate for the various steps in the CSP protocol, including an estimate of CPU time. Various heuristics are used to obtain the cost estimates of each stage in the CSP protocol. One important heuristic is that for the IEFF packing cost, it is assumed that the top 10 space groups will be sampled and that the cost for each space group is the same. When planning to sample more space groups this value can be scaled to get a minimum cost, however some space groups take longer to pack and optimize than others.
Note
Times reported in this Floe are CPU hours and not wall clock times.
This Floe can take any dataset as input (only a SMILES or 2D representation of the molecule is needed). The benzoic acid dataset in the “Psi4 QM Conformer Ensemble (Part I of CSP Protocol)” folder of the tutorial data is used below:
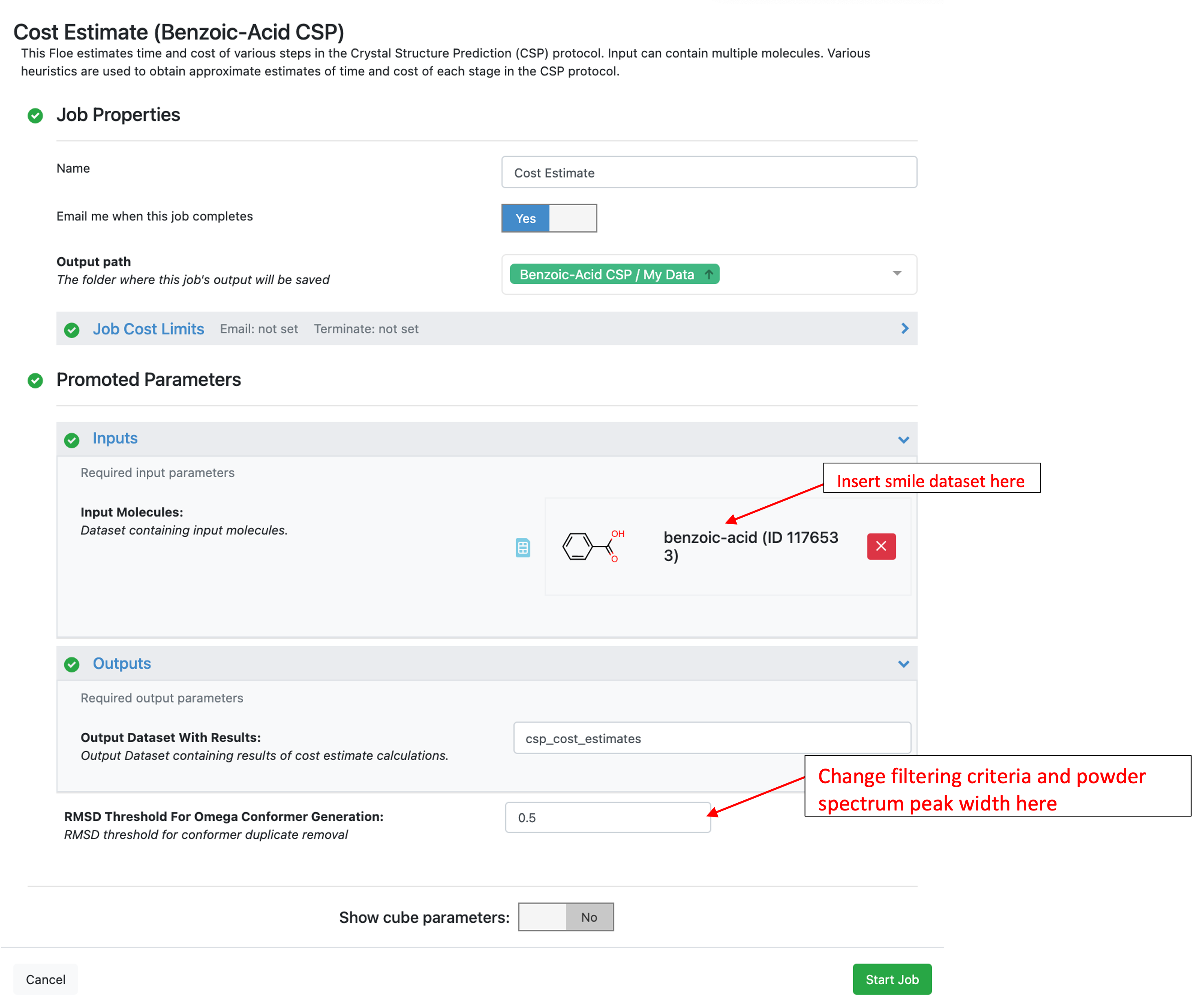
Here is the cost estimate report for benzoic acid: