Tutorial: Cheaper and Faster Version of Building Machine Learning Regression Models for Physical Property Prediction of Small Molecules¶
OpenEye Model Building is a tool to build machine learning models that predict physical property of small molecules.
In this tutorial, we will train several regression models to predict IC50 concentrations at which molecules become reactant to pyrrolamides. The training data is known as the Pyrrolamide Dataset. The results will be visualized in both the analyze page and floe report. We will leverage the floe report to analyze and choose a good model. Finally, we will choose a built model to predict the property of some unseen molecules.
This tutorial uses the following Floe:
Neural Network Regression Model Building.
Warning
This tutorial runs a barebone version of the floe, reducing parameters to a minimum. It builds 4 ML models, costs about a dollar and takes 10 minutes to run. We should not expect to build any good model in this version, but use it to understand the workings and/or a trial run. For full version refer to Building Machine Learning Regression Models.
Create a Tutorial Project¶
Note
If you have already created a Tutorial project you can re-use the existing one.
Log into Orion and click the home button at the top of the blue ribbon on the left of the Orion Interface. Then click on the ‘Create New Project’ button and in the pop up window enter Tutorial for the name of the project and click ‘Save’.
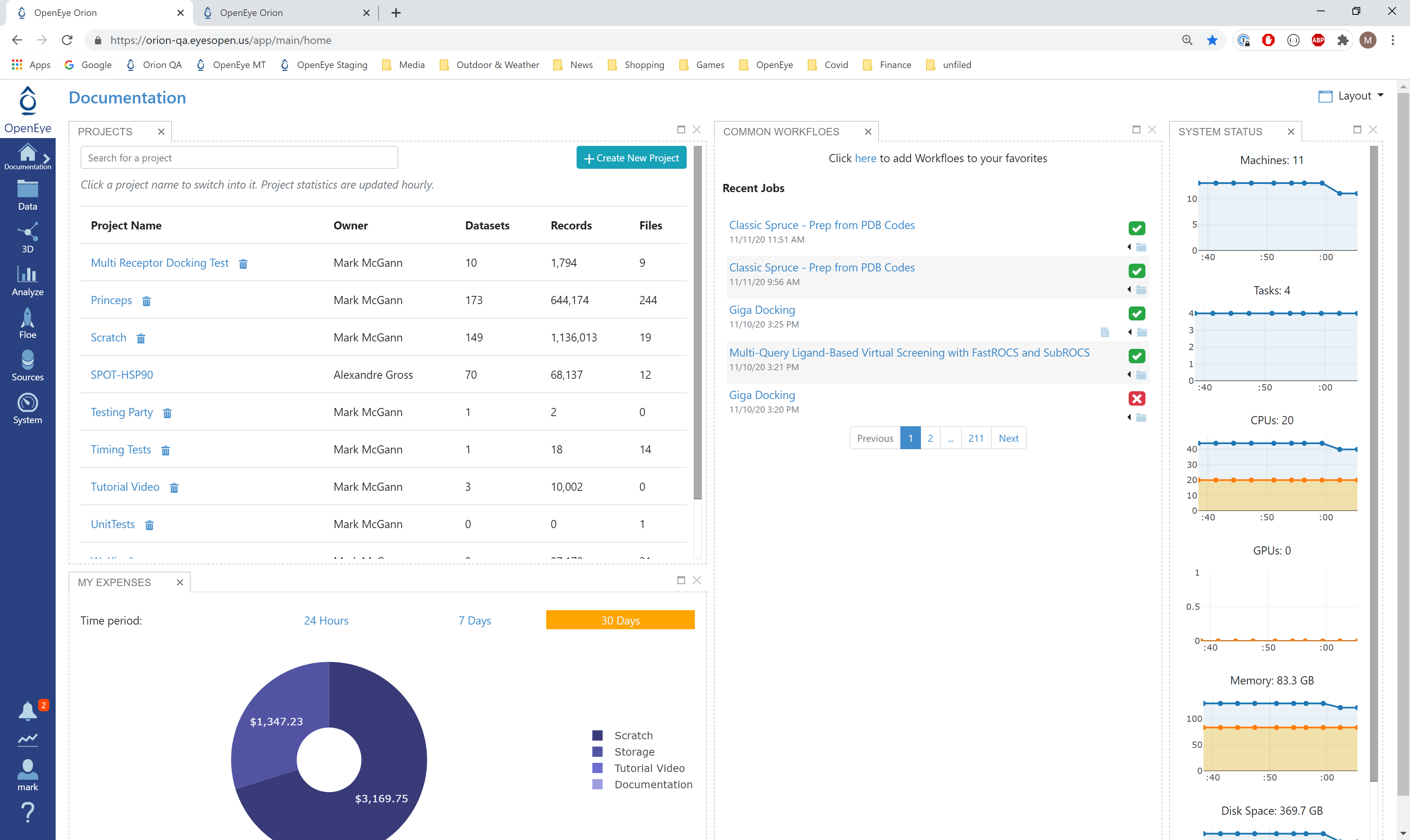
Orion home page¶
Floe Input¶
The Floe requires an input dataset file with each record in the file an OEMolField.
There needs to be a separate field in each record containing the Float physical property to train the network on. This field needs to be selected in the promoted field parameter Input Small Molecule Dataset For Prediction.
The Pyrrolamide dataset an contains several OERecord (s). The floe expects two things from each record:
An OEMol which is the molecule to train the models on
A Float value which contains the regression property to be learnt. For this example, it is the IC50 permeability concentration.
Here is a sample record from the dataset:
OERecord (
*Molecule(Chem.Mol)* : c1ccc(c(c1)NC(=O)N)OC[C@H](CN2CCC3(CC2)Cc4cc(ccc4O3)Cl)O
*NegIC50(Float)* : 3.04
)
You can try this tutorial on the following dataset:
Note
The input mol and training field can be filtered and transformed by the preprocessing units:
Promoted parameter Neg. Log Signal : Applies -Log10on training field
Promoted parameter Preprocess Molecule :
Keeps the largest molecule if more than one present in a record.
Sets pH value to neutral.
Applies the Blockbuster Filter
Run OEModel-Building Floe¶
Click on the ‘Floes’ button in the left menu bar
Click on the ‘Floes’ tab
Under the ‘Categories’ tab select ‘OpenEye Model Building’ package
In the search bar enter ML Build
A list of Floes will now be visible to the right
Launch the floe ML Build: Regression Model with Tuner using Fingerprints for Small Molecules and a Job Form will pop up. Specify the following parameter settings in the Job Form.
A list of Floes will now be visible to the right (see below)
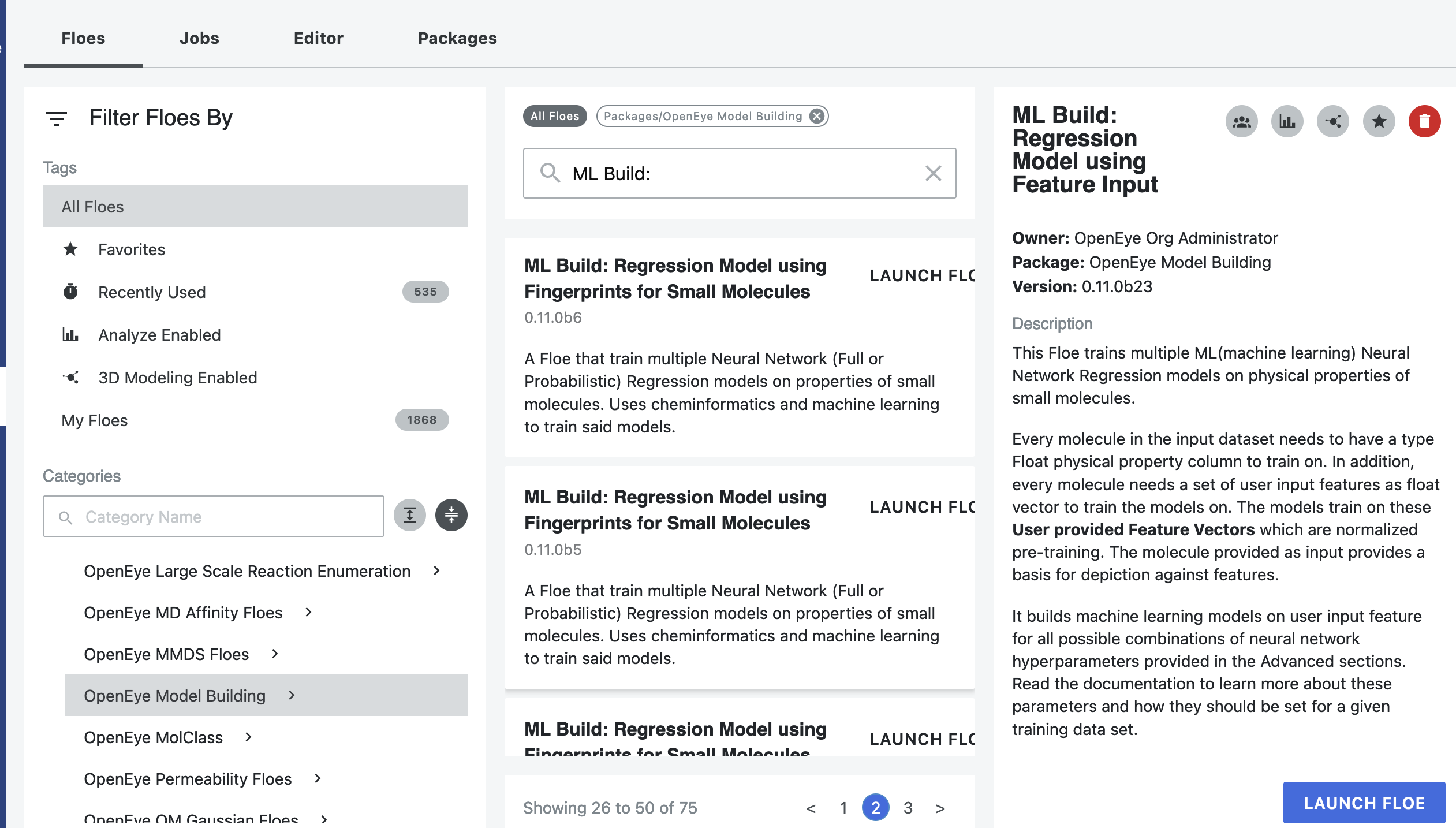
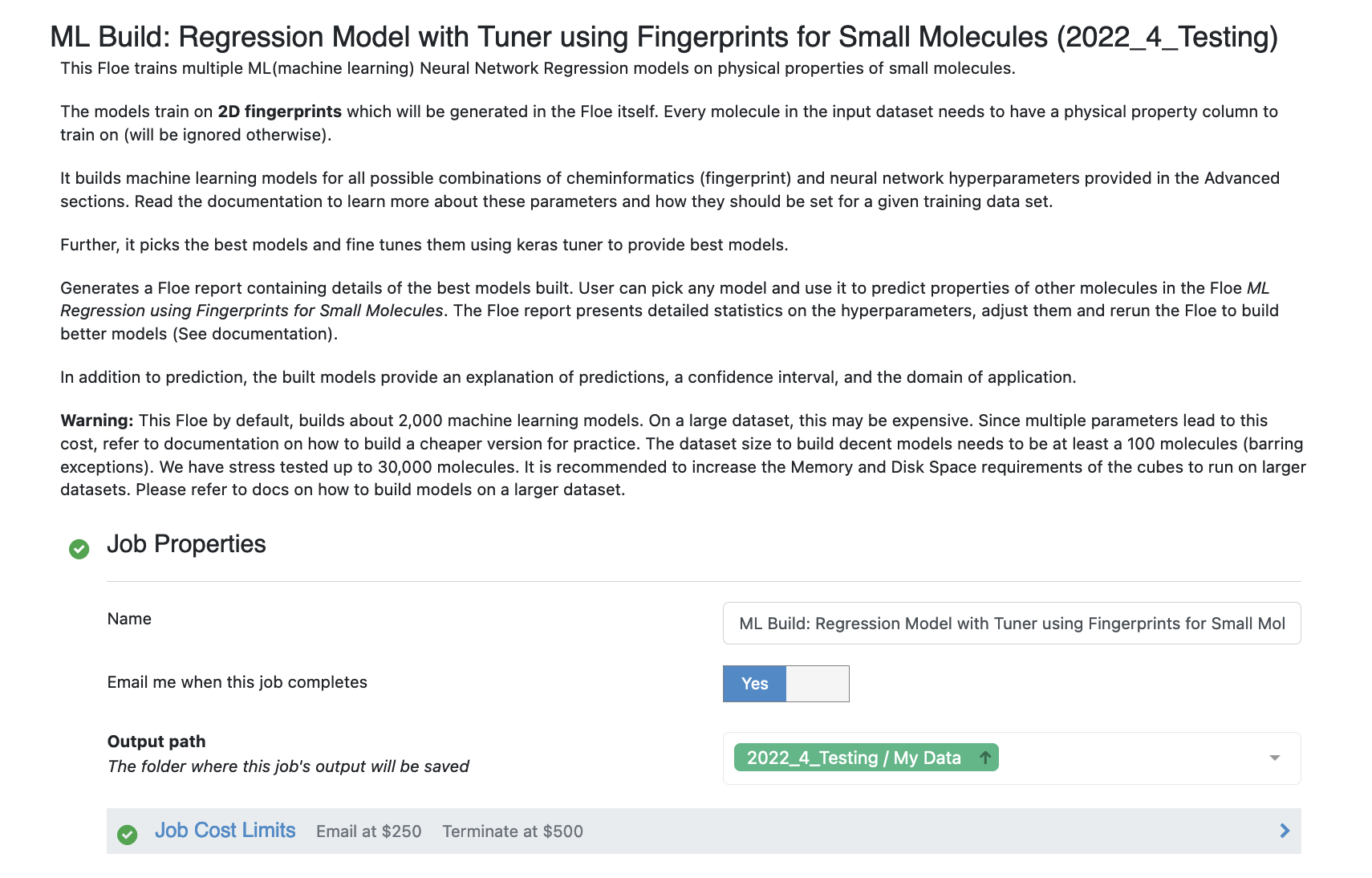
Click on the Small Molecules to train on button
Select the given dataset or your own dataset.
Post selecting the dataset, select the response value which the model will train on. This field dynamically generates a list of columns to choose from based on the upload column. For our data, its NegIC50.
Select how many model reports you want to see in the final floe report. This fields prevents memory blowup in case you generate >1k models. In such cases viewing the top 20-50 models should suffice.
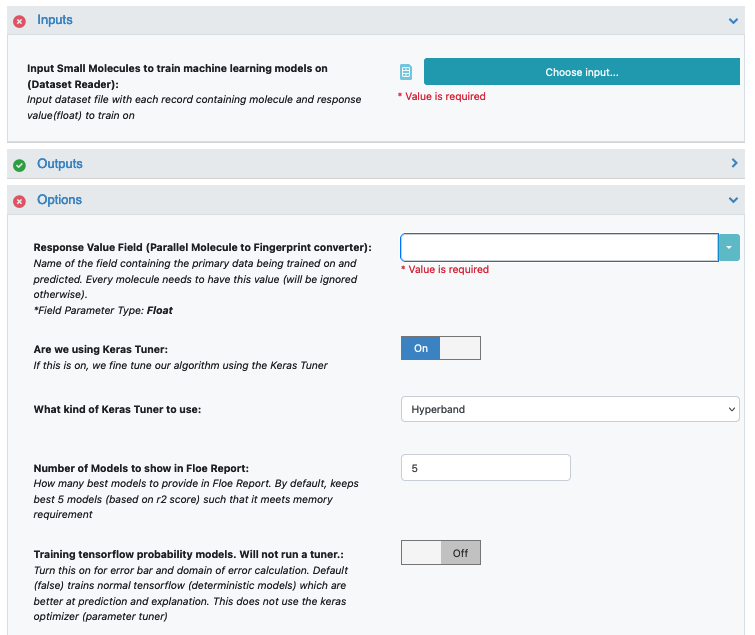
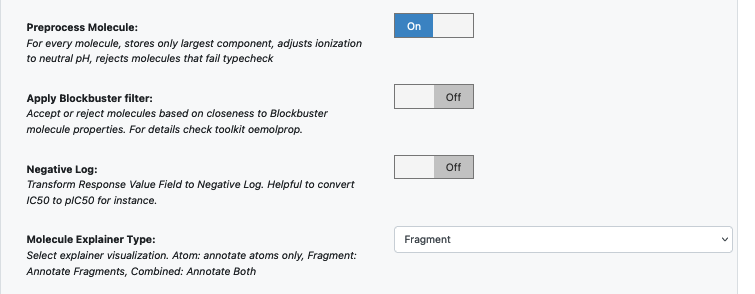
Under the Options tab, turn on the Boolean parameter Preprocess Molecule. This preps every molecule by removing salts, charge and multiple components, keeping the largest molecule. It also applies the blockbuster filter and sets the molecule to neural pH.
Select how you want to view the molecule explainer for machine learning results.
‘Atom’ annotates every atom by their degree of contribution towards final result.
‘Fragment’ does this for every molecule fragments (generated by the oemedchem tookit) and is the prefered method of med chemists.
‘Combined’ produces visualisations of both these techniques combined.
In case it is necessary to transform the training values (Pyrrolamides in this case) to negative log, turn the Log. Neg Signal switch as well. Turn it on for this example.
You can let the model run at this point and it should run with default parameters. But we can tweak a few parameters to learn more about the functionality.
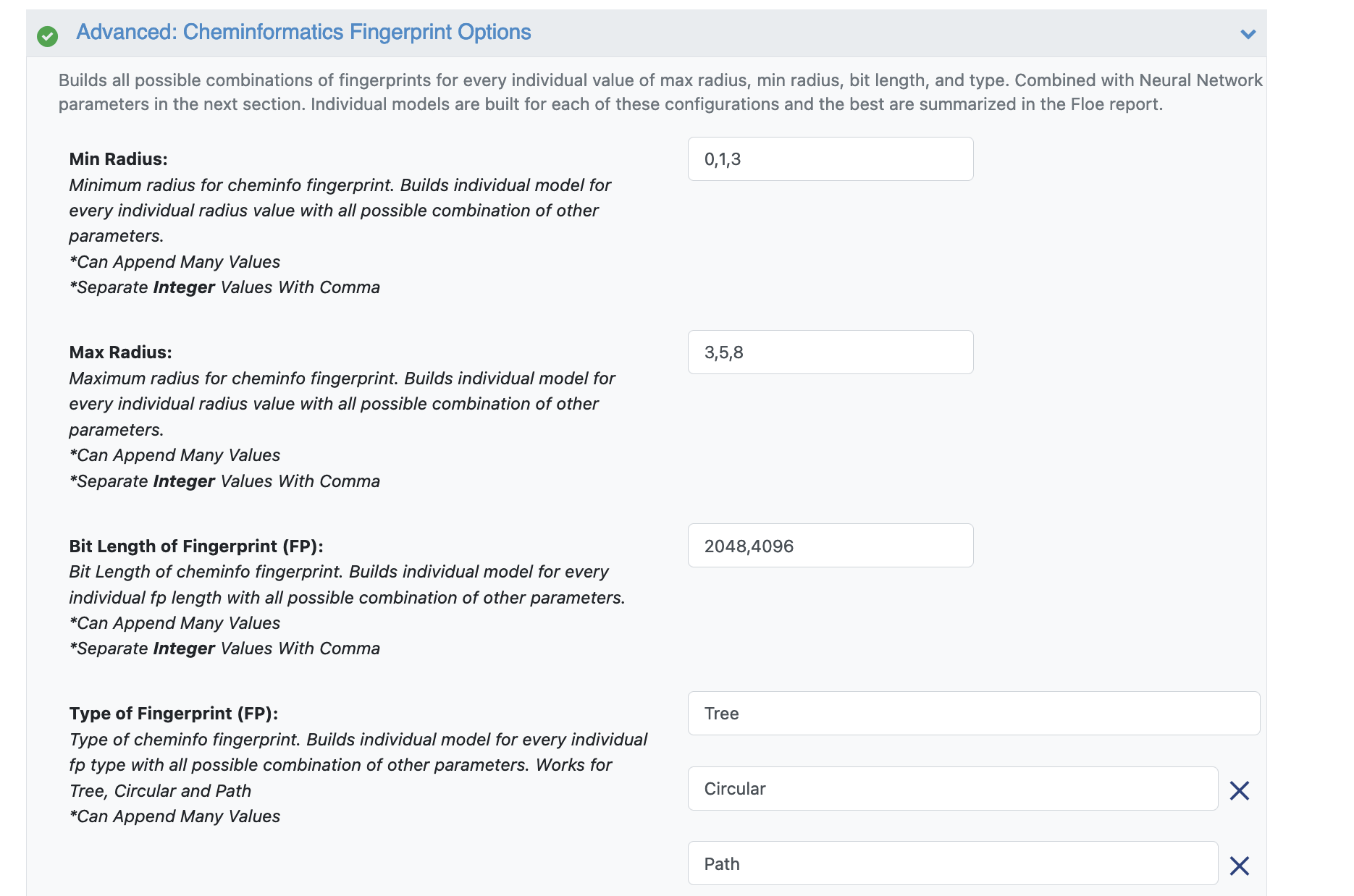
Open the Cheminfo Fingerprint Options. This tab has all the cheminformatics parameters the model will be built on. For this cheap and quick run of the floe, change the following:
Min Radius: 0
Max Raduis: 5
Bit Length of FP: 1024
Type of FP: Remove Tree and Path and keep Circular. (Or keep any one of them)
Note: the plots corresponding to these features in the floe report will now contain only a sigular point with no information on their variance
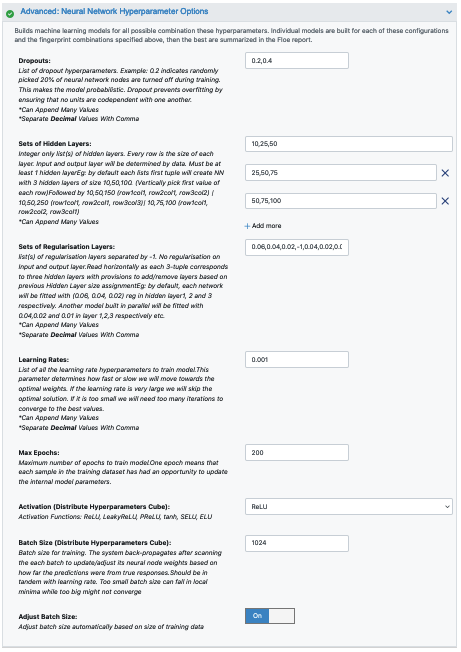
Next we move to the Neural Network Hyperparameters Options. This is where the Machine Learning parameters are listed. Again, for this cheap and quick run of the floe, make the following modifications:
Dropouts: 0.2
Sets of Hidden Layers: 100,50,0,-1,20,0,0
Sets of Regularisation Layers: 0,0,0,-1,0.06,.04,0.02
Batch size: 64
That’s it! Lets go ahead and run the Floe!
Analysis of Output and Floe Report¶
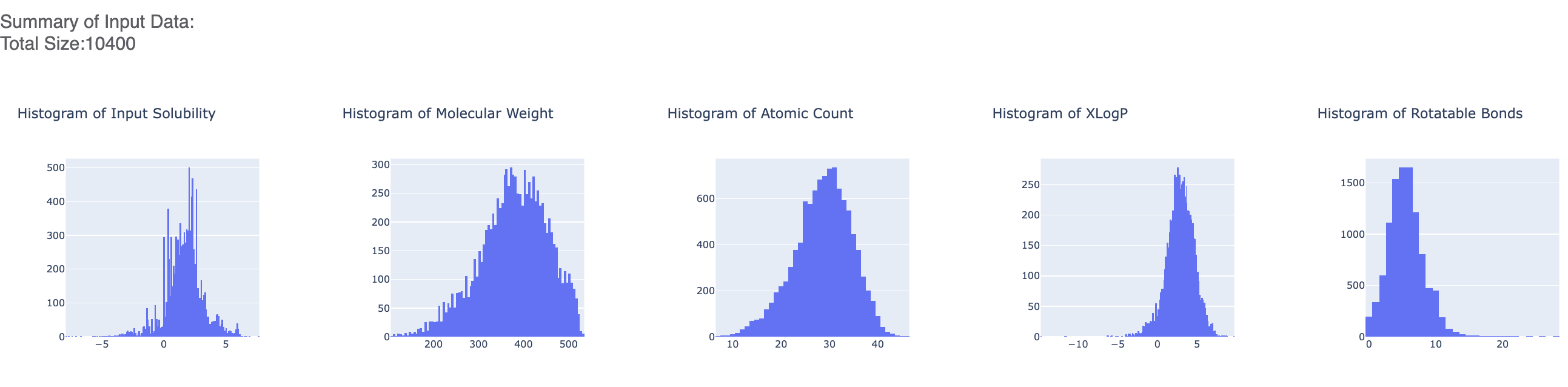
After the floe has finished running, click the link on the ‘Floe Report’ tab in your window to preview the report. Since the report is big, it may take a while to load. Try refreshing or popping the report to a new window (located in the purple circle in the image below) if this is the case. All results reported in the Floe report are on the validation data. The top part summarizes statistics on the whole input data. For this run, it should mostly be a singular point as we have just one values for most parameters.
The graphs show the Mean Absolute Error (MAE) for different values of the neural network hyperparameters. It helps us analyze how sensitive the different hyperparameter values are and plan future model builds accordingly. For instance, graph below shows that dropout of 0.1 and maxr of 5 are better choice for parameters in future model build runs.
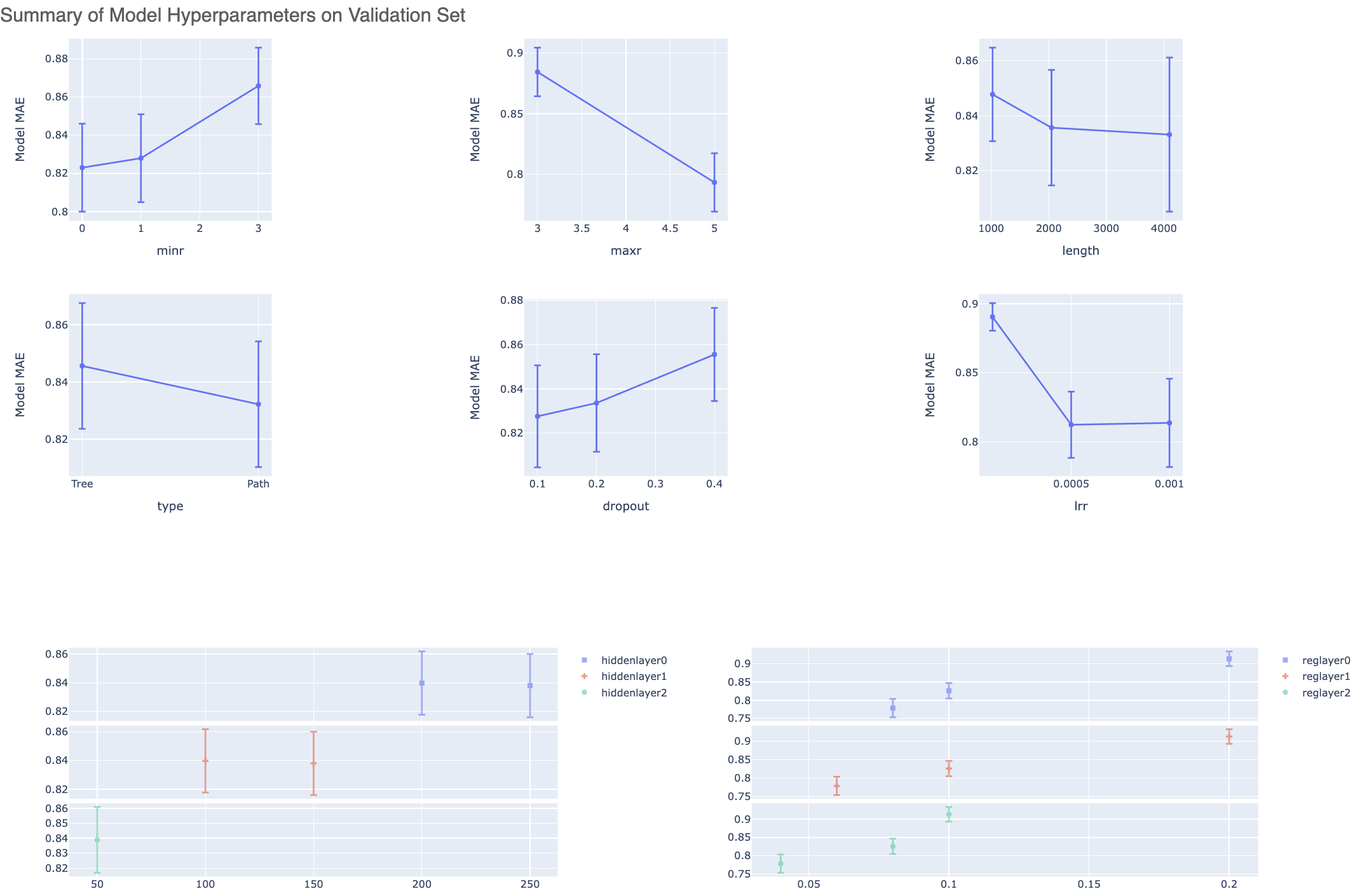
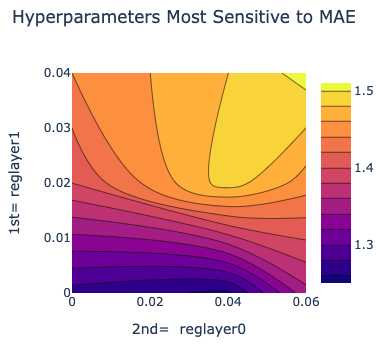
There is also a plot between the top two most sensitive hyperparameters. For our case, this graph may be empty due to lack of data.
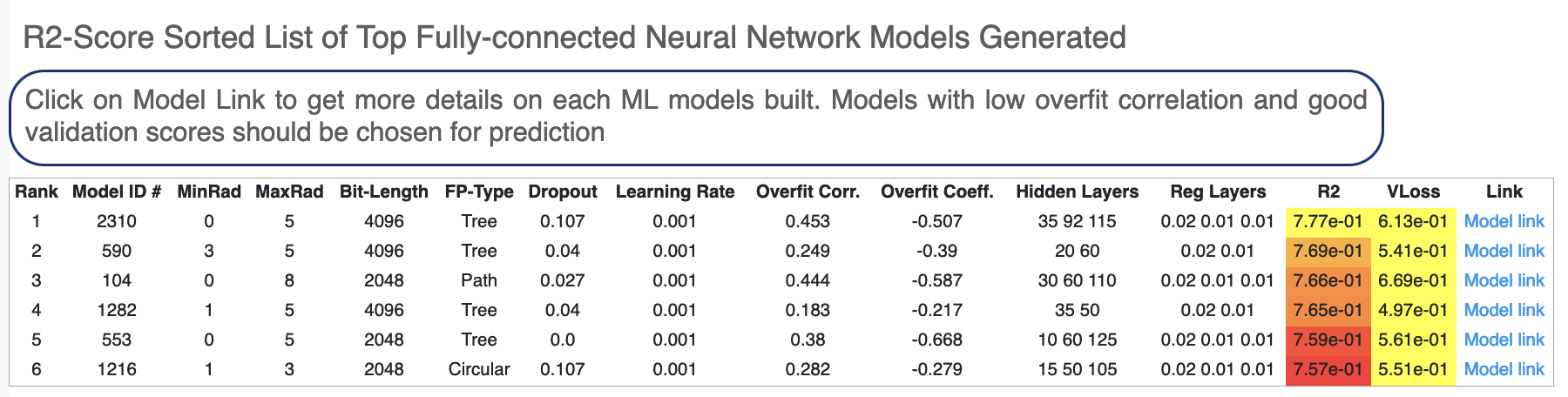
Next, we tabulate the list of all models built by the fully connected network. These models are sorted by the least R2 Score (for validation data). On the face of it, the first row should be the best model since it has the least error on the validation data. But there can several other factors besides the best R2 to determine this, starting with Loss in the next column. Hence, lets look at a sample model by clicking the ‘Model Link’. Let’s click on a model link and look at the training curves under ‘Neural Network Hyperparam and Fingerprint Parameters’.
For each model, we have a linear color scale showing the rank. We also have the cheminformatics and machine learning parameters the model was trained on.
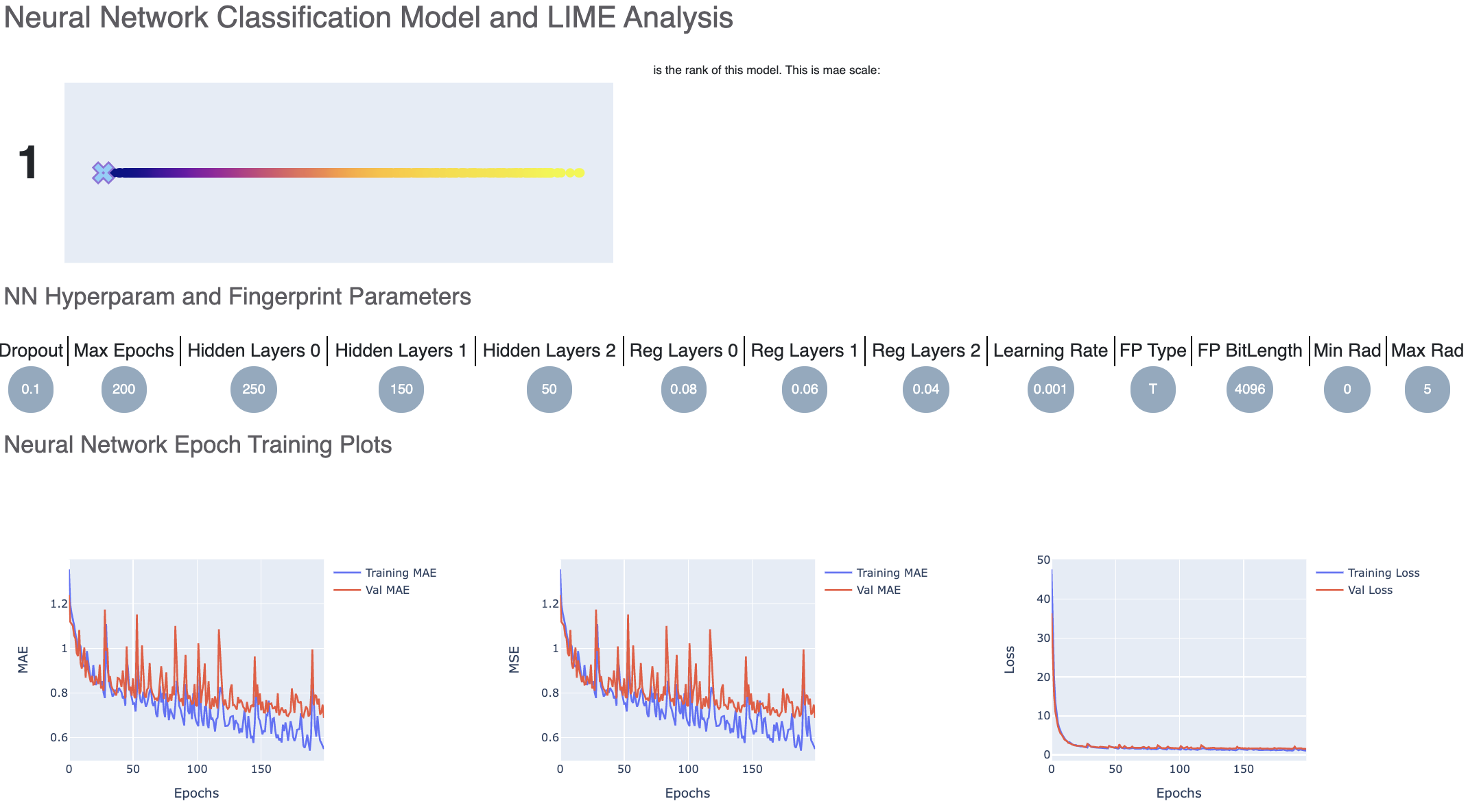
We see that the training and validation MAE follow similar trend which is a good sign of not overfitting. Had they been diverging, we might have to go back and tweak parameters such as number of hidden layer nodes, dropouts, regularizers etc. Although too much fluctuation in the graph suggests we need a slower learning rate.
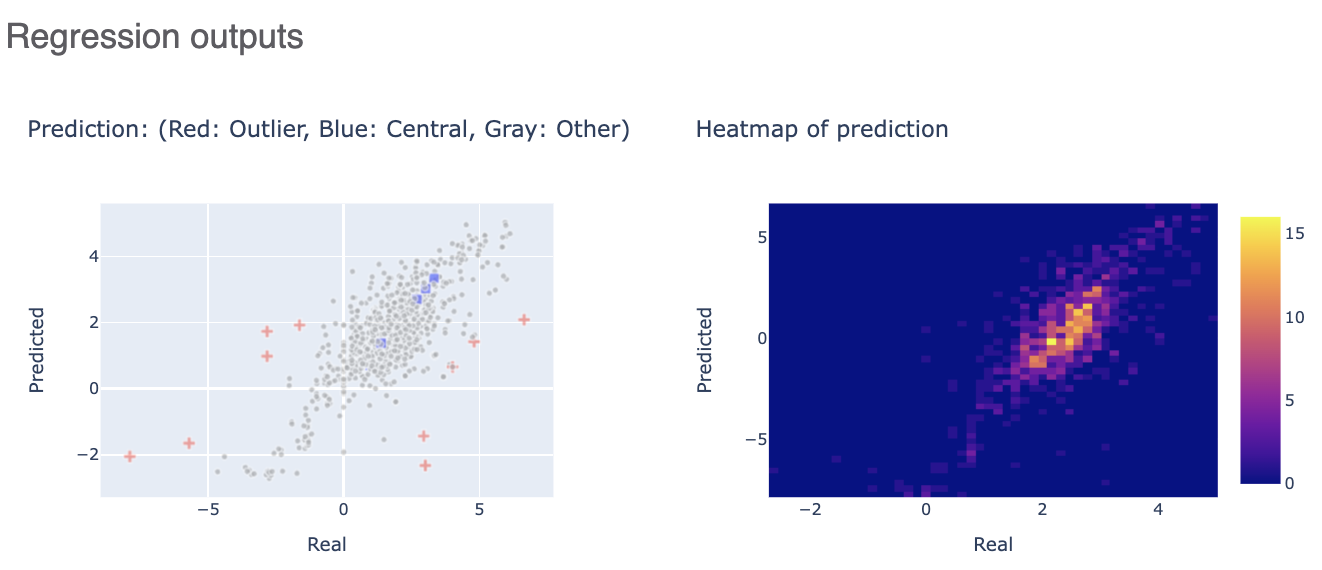
The Regression output plots real versus predicted to show how well the training is correlated. Below that, click on the interesting molecule to see the annotated explaner of the machine learning model.
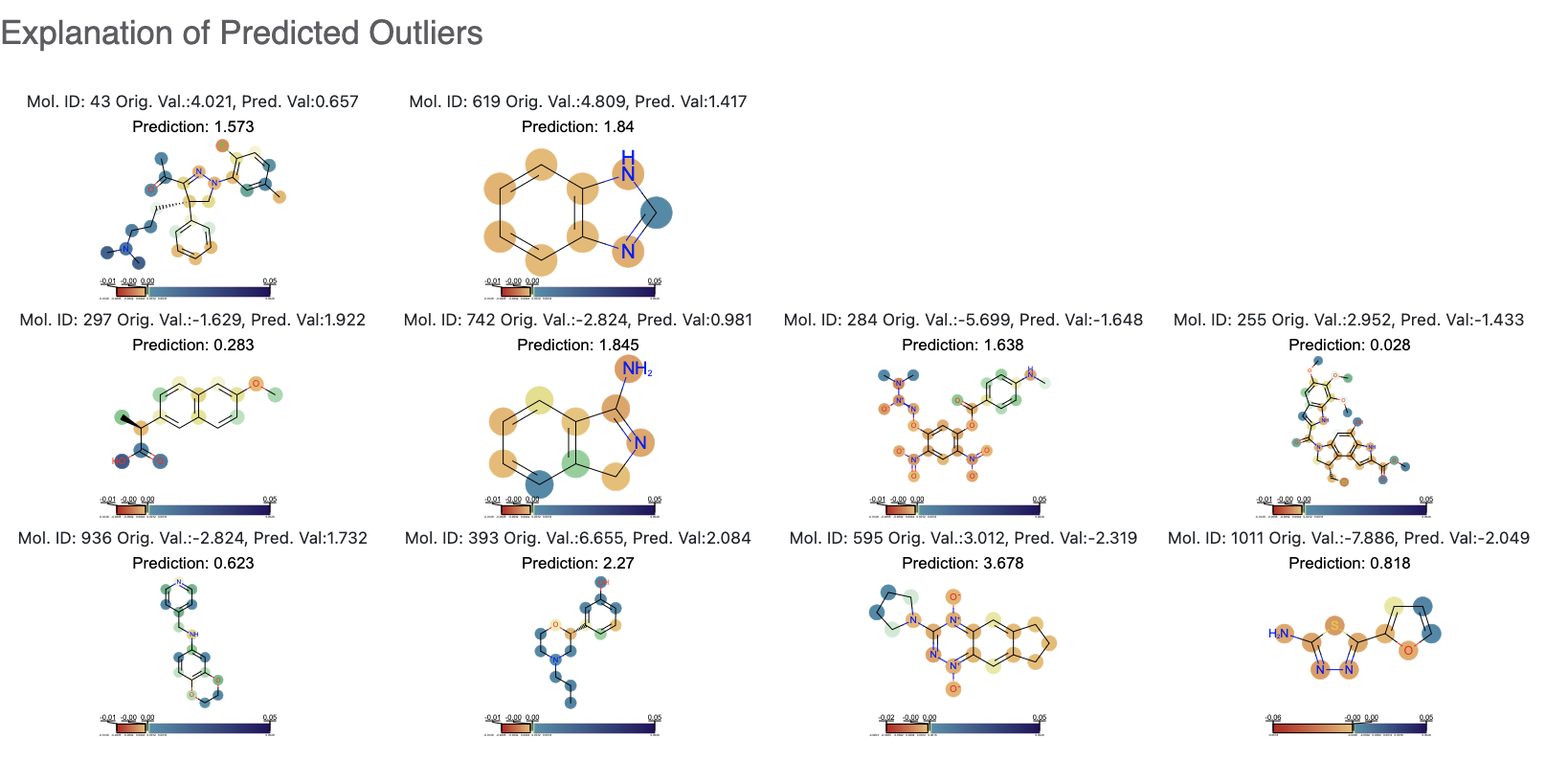
While this tutorial shows how to build a few regression models, building a usable ones is a non-trivial task. Refer to the original tutorial for this.
After we have found a model choice, we can use it to predict and or validate against unseen molecules. Go to the next tutorial Use Pretrained Model to predict generic property of molecules to do this.