Aligning Molecule Based on MCS
A program that aligns molecules based on their maximum common substructure.
Command Line Interface
A description of the command line interface can be obtained by executing the program with the –help argument.
prompt> python mcsalign2D.py --help
will generate the following output:
Simple parameter list
input/output options :
-fit : Align filename
-out : Output filename
-ref : Ref filename
Code
Download code
See also
Molecule Alignment chapter
OEIs2DFormatfunctionOEPrepareDepictionfunctionOEMCSSearch class
OEPrepareAlignedDepictionfunction
Example
prompt> python mcsalign2D.py -ref ace1.ism -fit ace2.ism -out out.mol
taking the output coordinates and generating the images with the mol2img program will produce the images shown in Table: Example of using the program to align two molecules based on MCS.
reference molecule |
aligned molecule |
|---|---|
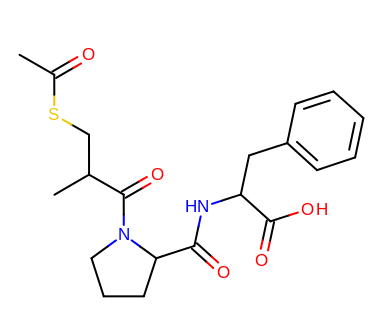
|
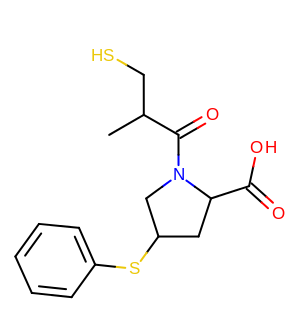
|
See also
Molecule Alignment chapter