Using RotFit
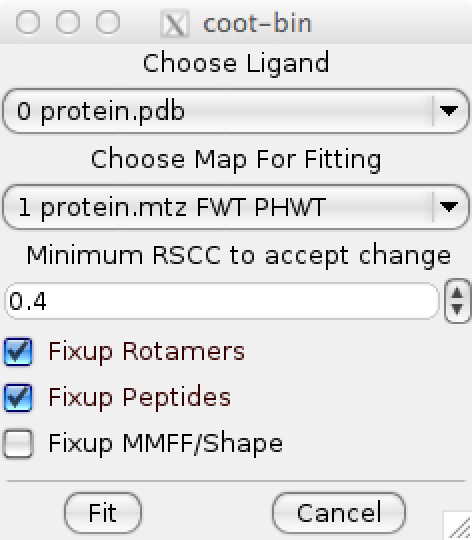
Activating the “Fit Sidechains…” menu option opens up the ROTFIT dialog:
Writedict Dialog
To use the ROTFIT dialog, a protein and mtz map must be loaded into coot. The ROTFIT interface deduces the appropriate regular and difference map to use and currently cannot handle non standard or unique column names.
ROTFIT options
ROTFIT is a program that fits protein side-chains into the surrounding density. It utilizes both regular and difference density to place side-chains appropriately searching both rotamers and peptide flips in addition to an optional full MMFF94-driven optimization into density.
Fixup Rotamer
Rotamer fixups selects from the probable rotamer sets to find the rotamer that best matches the surrounding density.
Fixup Pep-flip
Peptide flips are enumerated and the one that best matches the surrounding density is selected.
Fixup MMFF/Shape
Full MMFF/Shape minimization is performed on compatible residues attempting to fit the residue into the locally surrounding density.