Color Features
In addition to shape-alignments ROCS, optionally, considers chemistry alignment, known as ‘color’. User specified definitions of chemistry can be included in the superposition and similarity analysis process to facilitate the identification of those compounds that are similar both in shape and chemistry.
Color atoms are described as Gaussians and displayed in vROCS as colored spheres. The Gaussian for a color atom is relatively hard with a steep gradient. Figure: Hard vs. Soft Gaussians illustrates hard vs fuzzy Gaussians. Both Gaussians in the figure represent the same volume as the sphere. However, the hard Gaussian, with the steep gradient, reaches a probability of zero (0) within the radius of the sphere. The color features are either matched, if they fall within the sphere radius, or not matched. In the case of the fuzzy Gaussian there are areas outside the volume of the sphere (the area under the curve indicated by the two arrows) where the Gaussian probability is greater than zero. This would allow color features to match even when they align well outside the sphere representing the color atom. That situation would lead to less precise alignments and, for that reason, the ‘hard’ Gaussian is employed.
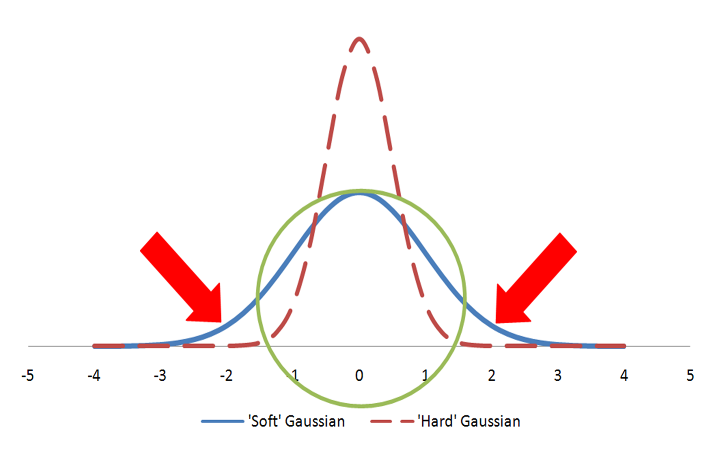
Hard vs. Soft Gaussians
A sphere described by two different Gaussian functions. The ‘hard’ Gaussian (dashed) is the one employed by ROCS to approximate a color atom sphere.
ROCS comes pre-loaded with two color force fields, Implicit Mills Dean (default) and
Explicit Mills Dean. These are described in color force field files (*.cff) located
in the ROCS data directory. A sample color force field file, sample.cff,
is also provided as a template for a user’s own custom force field. The desired force
field file is supplied to ROCS either at the command line using the -chemff command
or in the vROCS Preferences menu. Further information on editing color force field
files is given in the section Color Force Field.
The color force field is used to measure chemical similarity between the query and the database molecule and to refine shape-based overlays. The color force field file describes:
Color atom types
Which functional groups the color atoms should be applied to. ROCS uses only the heavy atoms of molecules, hydrogens are ignored.
Whether the interaction between color atoms is attractive or repulsive. Interactions between color atoms of the same type are always attractive. The weight term describes the strength of the interaction, relative to the shape gradients and the range term affects the range of the interaction.
The color features described in the Implicit and Explicit Mills Dean color force field files include:
- Donor:
Functional groups that can act as H-bond donors e.g. acid-OH
- Acceptor:
Functional groups that can act as H-bond acceptors e.g. carboxylate
- Anion:
Functional groups with either localized or delocalized negative charge e.g. tetrazole
- Cation:
Functional groups with either localized or delocalized positive charge e.g. guanidinium
- Hydrophobe:
Terminal or nonterminal aliphatic groups, including Br and I
- Rings:
Rings of defined size e.g. 4-7 atoms
A custom force field file can include other features that you define e.g. positive, negative, carbonyl_linker, metal_binder. For each color atom type a set of SMARTS is used to define the specific functional groups to which the color atom will be applied. The Implicit and Explicit Mills Dean force fields differ in these functional group definitions. For example, the Explicit Mills Dean force field allows a primary amine to be an acceptor as well as a donor whereas it is a donor only in the Implicit Mills Dean force field.
The color force field can also be used for post-shape scoring either alone, e.g. ColorTanimoto and Color Tversky, or in combination with shape scores, e.g. TanimotoCombo and TverskyCombo. Some additional scores are available with color:
ScaledColor
ComboScore
ColorScore
These scores are defined in the section Report File.