Usage Notes
Minimum Requirements
At the minimum, FLYNN requires a density file and a the connection table of the ligand - usually derived from a supplied molecule file e.g. .pdb, .sdf, .mol2, .smi. FLYNN automatically searches the density for volumes of unmodeled density that are similar in size to the input ligand. Because of this it is highly recommended that a protein is also input to FLYNN. This makes the job of locating the ligand density less prone to false positives.
Based on analyzing several hundred protein/ligand combinations, the best way to use FLYNN for ligand fitting is described as follows (of course your mileage may vary):
Always use a protein:
The protein is used to identify modeled density where the ligand cannot be placed. Without this, FLYNN will identify many candidate locations for placing the ligand that will need to be analyzed.
Note
Working with data from Cryo-electron Microscopy (cryoEM): FLYNN searches for “blobs” in the density after masking out the protein – it is in these “blobs” that FLYNN attemps to place ligands. For users working with cryoEM data, this means that, e.g., un-modeled density associated with the phospho-lipid membrane in membrane-bound structures will be (mis-)identified as potential binding site(s) for ligands. Our recommendation would be to either use volume segmentation tools to construct a mask for the membrane and mask out this portion of the map prior to feeding the map into FLYNN; or, to simply ignore the ligands placed in to this region (though, if there are many extraneous “blobs” identified in the membrane region, this may cause the program to run for much longer than otherwise necessary). The same general note can be made for regions of the map associated with proteins or other cofactors in a complex that are unmodelled: these regions should be masked out of the map prior to analysis with FLYNN, if possible.
Use difference maps for X-ray crystallographic data when appropriate:
X-ray reflection data should be refined without a previous ligand conformation or generated without the ligand present. While in many cases, for X-ray data, the same result will be generated, if the ligand is fit to data refined with a previous ligand conformation, FLYNN may be biased towards the density modified by the pre-existing ligand conformation. Similarly, for cryoEM maps, structures used as input to FLYNN should be docked or refined into the map without a ligand present. If the input structure to FLYNN has a ligand present, the “blob” associated with that binding site/pose may be masked out of the map, and new prospective ligand binding poses may not be generated appropriately.
use fragment mode for fitting fragments
If you have an input cocktail, use the -fragment flag. This option simultaneously fits all the cocktails against density and sorts them by best fit.
Types of input ligands
Not all ligands can be successfully fit by FLYNN. Such types include large polypeptides or proteins, very flexible molecules, or simply molecules that contain atoms that are not present in the MMFF94s force-field.
Perhaps the most important property is rotatable bond count. Although FLYNN may be able to generate conformers for molecules with more than 20 rotatable bonds, the results of such an exercise would be dubious at best and should be taken with a grain of salt.
FLYNN will automatically check for unhandled atom types, but will gladly try and fit large molecules or proteins that contain many rotatable bonds usually with disappointing results.
Note
Dangers of PDB format for ligand input Many crystallography packages do not enforce writing out connection table or bond order information when outputting pdb files, or worse, strip them out. If bond distances are not correct FLYNN’s attempt to automatically assign bond orders and atom types can be suspect. To facilitate this, FLYNN includes a -precheck flag that can be used to verify that the input ligand is identified correctly and for validation of the automatic bond order assignments.
Stereochemistry Enumeration
Compounds that contain unspecified or ambiguous definitions of stereochemistry will be preprocessed before conformational sampling to explicitly enumerate stereochemistry. Input molecules that have three dimensional coordinates inherently have stereochemistry specified, but SMILES or two dimensional SD files may have atoms (R/S) or bonds (E/Z) for which the stereochemistry is unknown or unspecified. In the case where the stereochemistry supplied is suspect, FLYNN provides an option that will completely enumerate all stereochemical centers.
The resulting
ligands will be sorted based on their best fit to density regardless
of the stereochemistry. That is, the best solutions will percolate
to the top of the output. See -sortAllChiral for more details.
Refinement Dictionaries
Both FLYNN and its companion program WRITEDICT can generate refinement dictionaries. A refinement dictionary is needed to maintain proper ligand geometry when using external reciprocal space refinement programs such as REFMAC5 or CNS X/PLOR.
Refinement dictionaries are a list of geometrical constraints encoding used chemical bonding and molecular conformation information. They are used by various refinement packages to describe standard geometries and constraints that are used during the refinement process. The quality of the post refinement ligand conformations are directly related to the quality of the constraints [Vagin-2004].
FLYNN comes bundled with a refinement dictionary writer named WRITEDICT. WRITEDICT uses the MMFF94 forcefield to derive geometrical constraints for the input ligands or ligand-protein complexes. The output dictionaries enforce, as closely as possible, the input ligand’s geometries while allowing the refinement programs to modify the geometry as needed.
WRITEDICT also automatically detects covalent bonds in pdb files and inserts the appropriate PDB LINK records and covalent bond entries in the output refinement dictionary.
REFMAC5 refinement dictionaries
When outputting REFMAC5 refinement dictionaries, WRITEDICT writes out a .cif file and a .pdb file. The .pdb file is written out for two reasons:
Inconsistent Hydrogen Naming
Some applications write out hydrogen names incorrectly in ways that cause REFMAC5 or visualization programs like coot or WinCoot to be unable to associate the hydrogens in the refinement dictionary with hydrogens in the pdb file. In the worst case, REFMAC5 will crash entirely.
When necessary, WRITEDICT renames and renumbers hydrogens, if they exist, so that REFMAC5 won’t crash and so the .cif file is consistent with the .pdb file.
Covalent Bonds
WRITEDICT detects covalent bonds and outputs proper LINK records in the .pdb file and proper covalent constraints in the .cif file. Without using both outputs, REFMAC5 will not detect covalent bonds during refinement.
Known residues
When WRITEDICT detects a known residue, it may remap the atom names to be canonical with the known residue (this prevents REFMAC5 from failing to refine). These new atom names are saved in the .pdb file that WRITEDICT outputs.
Input Ligands with no residue information
When WRITEDICT analyzes a ligand with no residue information, it assigns the whole ligand to a single residue (by default UNL). If this is not the desired outcome, the ligand must be put into PDB or MOL2 format and the residues must be manually assigned.
It is highly recommended to use WRITEDICT’s generated .pdb file in conjunction with the .cif.
Looking up known residues
WRITEDICT has an internal dictionary of known residues. By default, known residues names are retained when the graph of the known residue is exactly the same as the input residue. When this occurs, writedict can replace REFMAC5’s dictionary with the MMFF94 generated dictionary. If the graphs do not match then WRITEDICT relabels the residue with a new residue name since a different dictionary needs to be created.
Options available for residue matching is as follows:
Option
Meaning
exact
Known residue and input residue graphs must match exactly.
substructure
Input residue may be a substructure of the known residue.
Cannot be used with the exact flag.
fuzzybonds
Bond orders do not need to match
atomname
Atom names must match
Options are set using the -strict flag and joined with colons (:).
The default is:
-strict substructure:fuzzybonds
To reject dictionaries that do not match exactly, use:
-strict exact
However, the more permissive default setting has been known to generate invalid dictionaries on occasion. It is always safe to force exact matches.
To have WRITEDICT attempt to see if the ligand’s residue has already been deposited in RCSB simply add the -lookup switch. This forces WRITEDICT to compare (based on the current -strict settings) the input residue to all known residues. This is useful if the input is from a 2D connection table, such as smiles or does not contain residue information.
MMFF94 versus MMFF94s forcefields
The MMFF94s forcefield is a variant of MMFF94 that emulates time-averaged structures typically observed during crystallographic structure determination, mainly planar geometries at unstrained delocalized trigonal nitrogen centers. However, there are many theoretical studies that show nitrogen centers are puckered [Halgren-VI-1999].
That being said, due to the prevalence of crystallographic examples where time averaging has occurred, many chemists erroneously consider the time-averaged structure to be correct, hence this variant is available for use.
WRITEDICT approximates the MMFF94s forcefield by enforcing planar aniline nitrogen configurations using the out of plane atom types and parameters from the MMFF94s. See the -planarAniline parameter to specify planar constraints.
FLYNN and writing CIF Files
When FLYNN is run, CIF dictionary files are always written as output. Because the dictionary files are not conformation independent, they are written as if the molecules were split into separate files whether or not the user specifies FLYNN’s -split flag. (See the split flag in Advanced Options below for more details). These files are numbered, however, so that the file with the lowest number corresponds to the best scored fit ligand.
For example, consider using the -sortBy plp flag and getting the following output:
1nhu-poses_n001_b001_s01_c003.pdb 1nhu-poses_n002_b001_s01_c000.pdb 1nhu-poses_n003_b001_s01_c001.pdb 1nhu-poses_n004_b001_s01_c002.pdb
This indicates that the best fit to density (c000) has the second best PLP score (n001). By default the files are sorted by RSCC.
FLYNN CIF output consists of the following files:
a CIF file
a PDB file (in case atom names were remapped) or links were added.
an OEB file that contains the original molecule with annotations
It is safest, however, to run WRITEDICT on the output ligand and protein complex. The CIF files generated by FLYNN are primarily useful for adjusting torsions and geometries in various ligand building programs, such as coot.
Results on The Gold Test Set
Included in the FLYNN distribution is a copy of the protein+ligand complexes that have structures factors available from the Electron Density Server [Kleywegt-2004].
Along with this data set is a simple python script that can be used to fit ligands to the test data set. OpenEye has prepared the input test set as follows:
The Structure Factors and protein complexes were downloaded from EDS.
The ligands were separated from the proteins.
The protein was re-refined using the original mtz file to remove ligand bias.
A simple directory structure was created as follows:
pdbcode/protein.pdb - the re-refined protein
pdbcode/ligand.pdb - the original ligand
pdbcode/protein.mtz - the re-refined difference map density.
The following python script was used to run FLYNN on all of the proteins. (This script is also included in the FLYNN distribution):
#########################################################################################
## Copyright (C) OpenEye Scientific 2007,2008,2009,2010,2011
#########################################################################################
## This script analyzes the Gold dataset shipped with the flynn distribution
import os,sys
command = "../../../bin/flynn -in %(CODE)s/ligand.pdb -map %(CODE)s/protein.mtz " \
"-prot %(CODE)s/protein.pdb -out %(CODE)s_results.sdf " \
"-verbose -rms -reportfile %(CODE)s.log %(REDIRECT)s %(CODE)s.out"
if sys.platform == "win32":
REDIRECT = "2>"
else:
REDIRECT = "&>"
RMS = []
for file in os.listdir("."):
print file
if os.path.isdir(file) and len(file) == 4:
code = file
CMD = command % {"CODE":code, "REDIRECT": REDIRECT}
print CMD
os.system(CMD)
index = None
rmses = []
for line in open("%s.log"%code):
if index is None:
index = line.strip().split(",").index("RMSD")
else:
rmses.append( float(line.strip().split(",")[index]) )
if rmses:
print "\t===>Lowest RMS", min(rmses)
The results of FLYNN are shown in Figure Gold Test Set Results. The mean RMS to the crystal structure is 0.676 Angstroms.
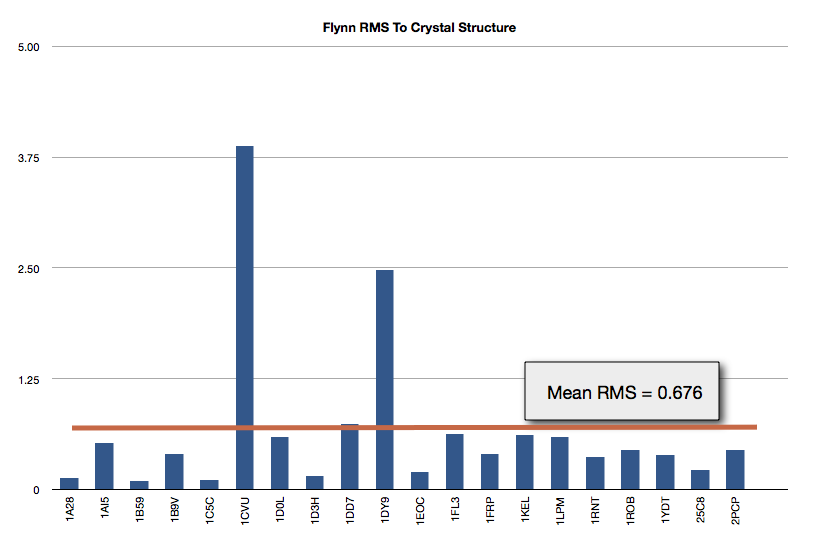
Gold Test Set Results