Search Buttons
There are a total of five search options.
Search Buttons
Analog Search
This is the default search, which is performed whenever a new molecule is loaded into the application. It attempts to find the best model compounds in the database by exhaustively searching in a chemically aware manner. Firstly if there is an exact match in the database the measurements associated with that molecule are reported first. Then the application performs a rooted maximum common subgraph search beginning at each ionizable region, and applies a positive score for matches and a penalty for substitutions, graded based upon the electronic commutative distance to the ionizable atom. The sum of these scores for each ionizable region match determines the overall rank of the molecule and its measurements. Only molecules with measurements that pass the property filters will be reported back. If you do not wish this search to be performed every time you load a new molecule simply turn that option off in the preferences.
Exact Search
This search attempts to find an exact match in the database, and reports back only those measurements which pass the property filters.
Property Search
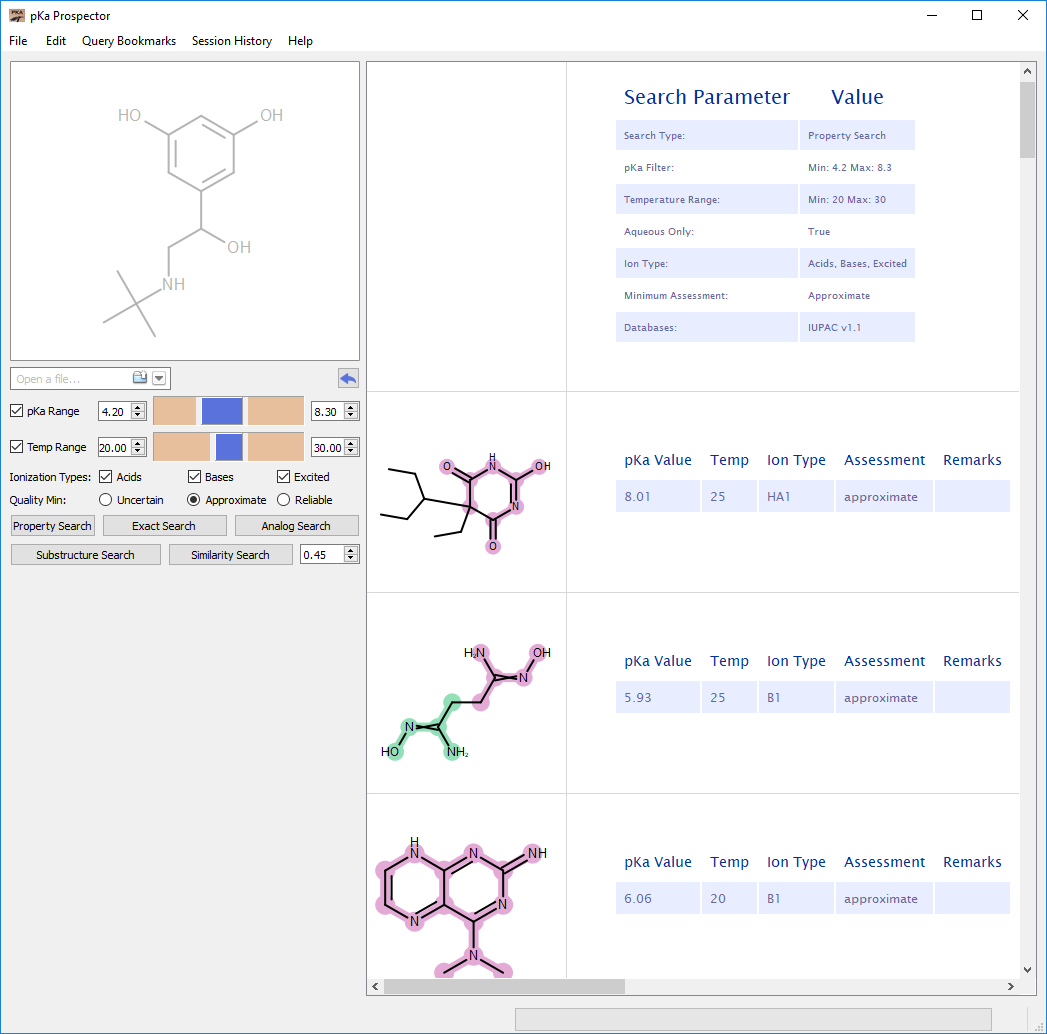
Property Search
This search ignores any query molecule information and simply reports back those molecules which pass the property filters. The query molecule depiction is deemphasized, and the results view does not have a query molecule in the first row. Figure: Property Search
Substructure Search
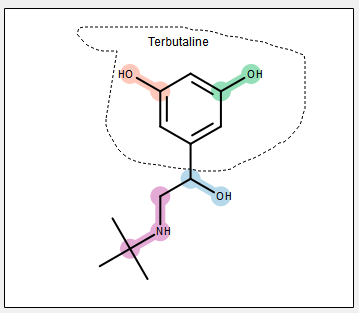
Lasso a Substructure
This search will look for molecules that have a common substructure with the query molecule. A subset of the query molecule can be used by lassoing with the mouse. Figure: Lasso a Substructure Again only molecules with measurements that pass the property filter will be reported.
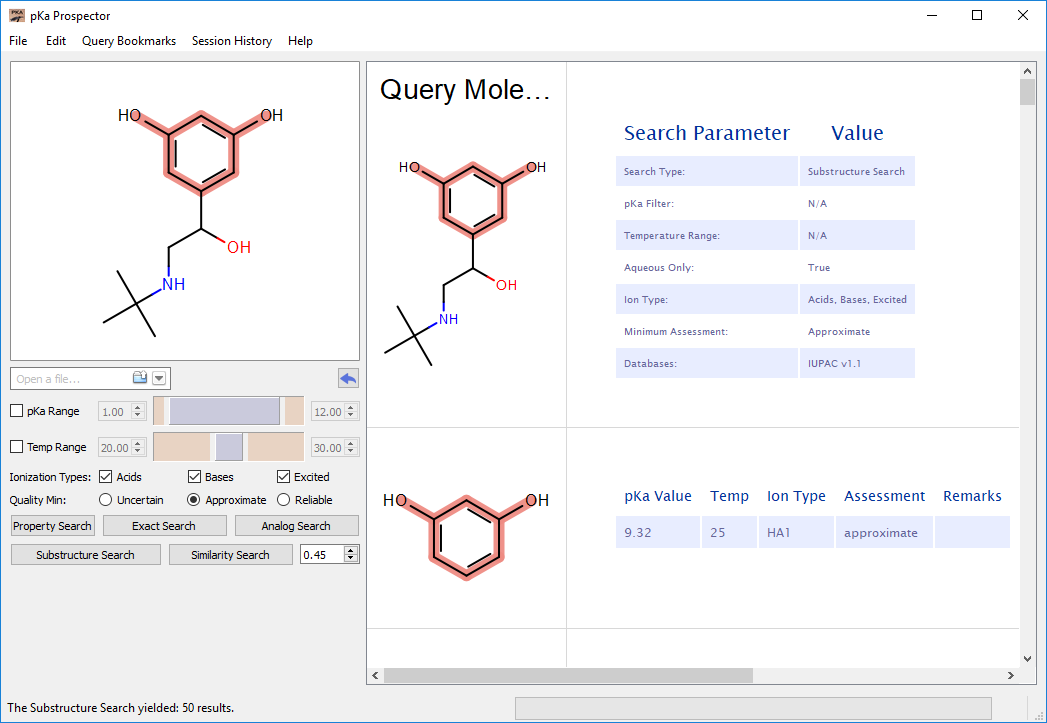
Substructure Search
Similarity Search
This search will perform a circular fingerprint search which is generated by exhaustively enumerating all circular fragments grown radially from each heavy atom of the molecule up to a given radius and then hashing these fragments into a fixed-length bit vector. Only measurements which pass both the property filter and are above the Tanimoto cutoff value will be reported.