Example Commands
Grid Calculations
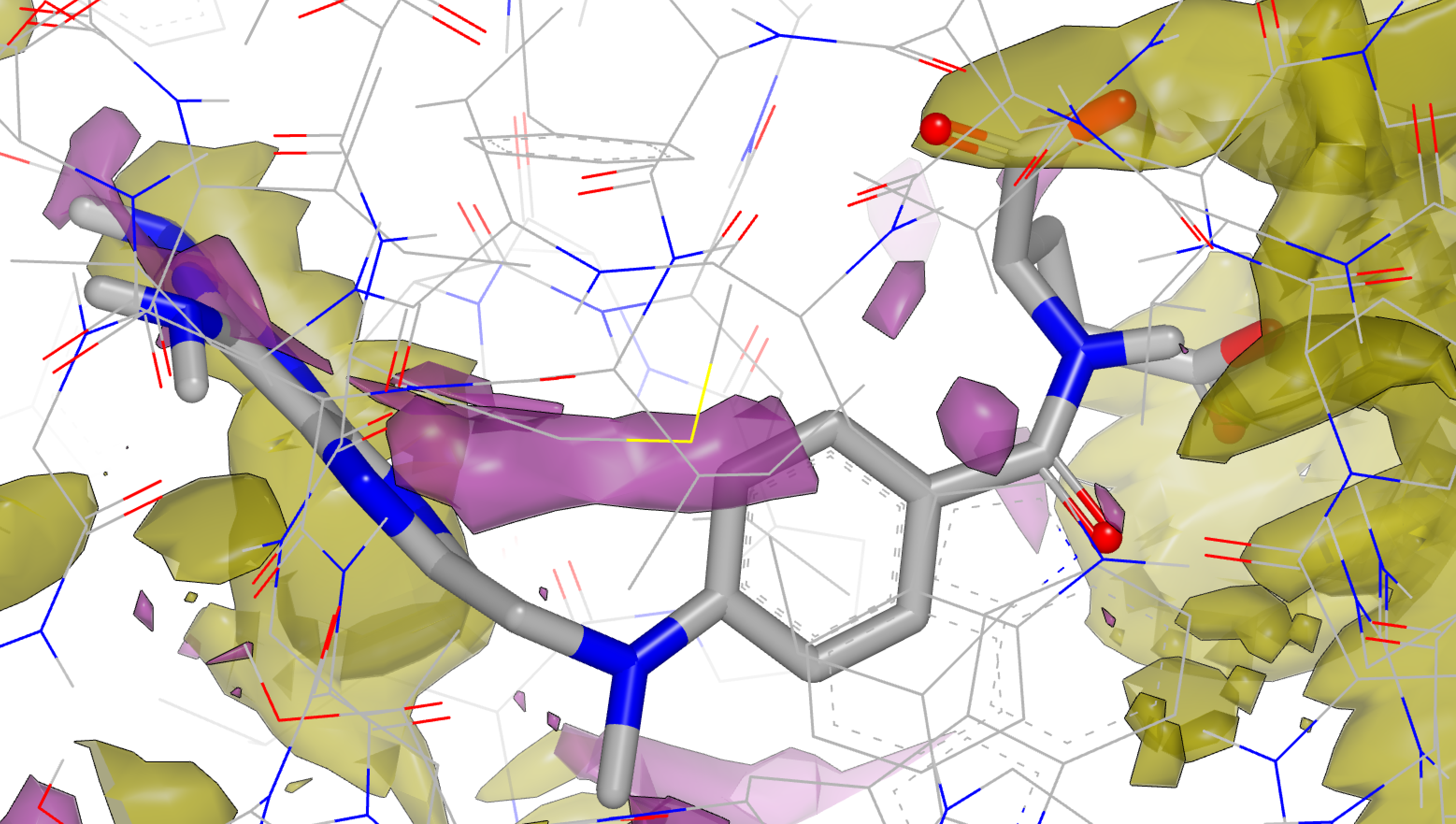
apo Neutral Difference Free Energy Grid
If a ligand is provided, either bound in the OEDesignUnit or separately on the command line, and no other selection criterion are specified, SZMAP will perform grid calculations centered around the ligand. If not using a design unit as input and the structure has multiple binding sites, it is usually appropriate to focus on only one of these by making sure only one molecule is in the input ligand file. Other ways to define the calculation area include a bounding box, the region around a particular protein residue or small molecule, even the entire surface of the protein. Beware, surface calculations generate a very large number of points and take a correspondingly long time to compute. It is usually best to focus the computation on a smaller region.
> szmap -mpi_np 4 -stbl -prefix 1dds_stbl -p 1DDS_AB__DU__MTX_A-201.oedu
or
> szmap -mpi_np 4 -stbl -prefix 1dds_stbl -p 1DDS_AB__DU__MTX_A-201_target.oeb.gz -l 1DDS_AB__DU__MTX_A-201_ligand.oeb.gz
Because there are often a great many points to be tested in a grid calculation, the
MPI multiprocessing options -mpi_np <num> and -mpi_hostfile <file>
can be used to speed calculations by running SZMAP on multiple cores
or multiple networked computers.
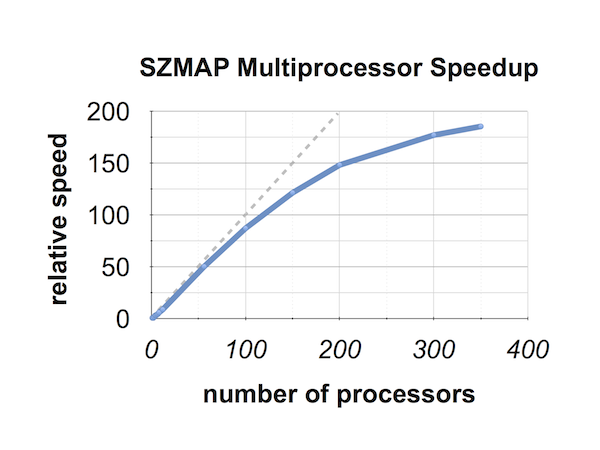
Speed vs Processors
The figure Speed vs Processors is from a stabilization grid calculation run across a wide range of processor numbers. The dashed line is the diagonal for reference. With 8 processors, SZMAP runs over 6 times faster, and with 200 processors, about 150 times faster.
By default, grid output consists of four types:
neutral difference free energy, a hydrophilic/hydrophobic polarity scale described below,
van der Waals energy, an energy term not included in the neutral difference free energy,
order, the fractional loss of rotational entropy due to electrostatics, indicating how much energy can be liberated by displacing water at this site,
mask, an accurate solvent accessible surface—the region where the calculations were performed and the probe did not clash.
Neutral difference free energies compare the water probe with an uncharged or neutral water probe, a water where all the atoms have a partial charge of 0.0. The uncharged probe is a proxy for a hydrophobic group of the same size and shape as the standard probe water and these difference energies map out variations in the hydrophobic/hydrophilic nature of the binding site. High affinity ligands put nonpolar groups where these energies are positive and polar groups where these energies are negative, mimicking the donor-acceptor pattern of the solvent in the apo pocket.
Neutral difference free energies do not capture variations in van der Waals energies, which subtract out in the difference calculation. Van der Waals results are provided separately and complement the information in the neutral difference free energies.
Many additional grids are produced when the option -results_set max is specified.
The most useful of these extra grids are probably psolv and wsolv—protein and water desolvation,
respectively. They are always penalties (> 0.0 kcal/mol) and reveal
the extent to which burying a charge costs energy, which needs to be compensated for by
a complementary charge.
For most grid calculations, the -stabilization option is recommended. This causes
complex, ligand and apo calculations all to be performed
(although ligand calculations are by default restricted to solvent regions from the complex).
Stabilization free energy values—computed as (complex + bulk) - (apo + ligand)
where bulk-solvent is defined
as 0.0—show how water energies are affected by binding. Negative stabilization free energies
are regions that are stabilized by binding, or looked at another way, regions that
stabilize binding. The simplest thing to do with them is leave them as they are, since they
increase affinity. They can be displaced without destroying affinity, however, but only
if they are displaced with a group that makes similar beneficial interactions.
Positive stabilization free energies indicate regions where water
is destabilized on binding. These waters lower affinity and are relatively easy to displace productively.
Note that positive stabilization free energies can be found in regions where the
neutral difference free energies in the complex
are negative, and vice-versa. These two types of energies are not always
correlated.
When comparing a series of ligands, all docked into the same binding site,
the apo calculations can be done once separately by dropping the -stabilization option and
specifying -apo_grids. Then these apo grids can be
used in stabilization calculations for a series of ligands with the -use_apo_from option.
If the ligands are all concatenated into one file, the -split_lig option will split each one
out and perform the complex and ligand and stabilization calculations one-by-one.
> szmap -apo_grids -prefix 1dds_apo -p 1DDS_AB__DU__MTX_A-201.oedu -l 1dds_lig_series.oeb.gz
> szmap -mpi_hostfile myhosts.txt -stbl -use_apo_from 1dds_apo.oeb.gz -split_lig \
-prefix 1dds_stbl -p 1DDS_AB__DU__MTX_A-201.oedu -l 1dds_lig_series.oeb.gz
or
> szmap -apo_grids -prefix 1dds_apo -p 1DDS_AB__DU__MTX_A-201_target.oeb.gz -l 1dds_lig_series.oeb.gz
> szmap -mpi_hostfile myhosts.txt -stbl -use_apo_from 1dds_apo.oeb.gz -split_lig \
-prefix 1dds_stbl -p 1DDS_AB__DU__MTX_A-201_target.oeb.gz -l 1dds_lig_series.oeb.gz
Note
The line above with a ‘\’ as the very last character indicates that the command was too long to fit on one line and continues on the following line.
Most grids are masked to show only regions where the minimum Coulombic interaction + vdw energy is
at or below -mask_cutoff (0.0 kcal/mol by default); anything
above this is considered a CLASH. The mask_grid represents this solvent-accessible region.
Somewhat surprisingly, errors in coordinates of
atoms in a structure, even small errors, occasionally lead to clashes being indicated in areas where
a crystallographic water is observed. Typically, there will be an adjacent region where
the probe water does not clash.
Grid results can be processed with the utility GridComp to break-out the region of the apo grid that has been displaced by the ligand.
> grid_comp -op lig_disp -i 4std_stbl.oeb.gz -o 4std_stbl_disp.oeb.gz
The volume of water displaced by the ligand plus the volume of water in the complex equals the volume of water in the full apo pocket. Displacement grids are useful in identifying, for example, precisely which waters are displaced by each of a series of substituents.
“At Coords” Calculations
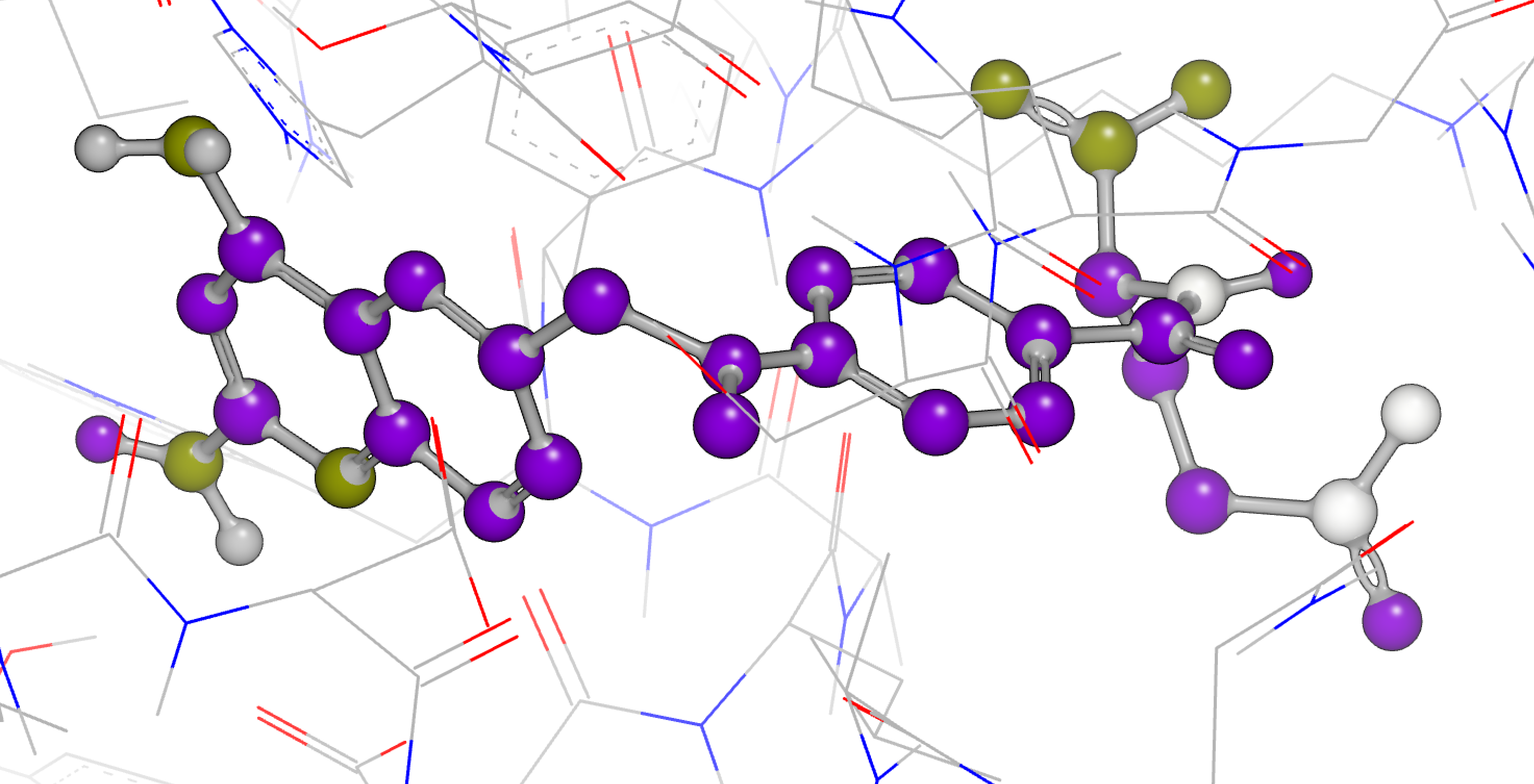
apo Neutral Difference Free Energy at Ligand Coordinates
When the -at_coords option is used, no grid is produced. Instead calculations
are done only at the coordinates of any atoms in the coordinates file and written out attached
to those atoms for use by SzmapReport, the Water Orientation VIDA Extension
and the Color By Atom Properties.
A tab-delimited table of these results is written to a text file, which can easily be
loaded into a spreadsheet program for further analysis.
“At coords” calculations produce many different results values at each point
(see the Appendix 2: SZMAP Atom Properties), but the four types of results enumerated
in the grid section above are the most useful.
> szmap -prefix 1dds_coords -p 1DDS_AB__DU__MTX_A-201.oedu -at_coords 1dds_lig.oeb.gz -excludeDUligand
> szmap -prefix 1dds_coords -p 1DDS_AB__DU__MTX_A-201_target.oeb.gz -at_coords 1dds_lig.oeb.gz
SZMAP -at_coords calculations are much faster than grid calculations
because they operate over a much more limited set of points. Keep in mind that a region
where there is a large gradient in a property will be undersampled if the coordinates there
are widely spaced. Points labeled “CLASH”
have Coulombic interaction + vdw energy greater than the -mask_cutoff
(analogously to how the mask_grid is determined for a grid calculation).
The “CLASH” label can be displayed in VIDA using the generic label function under the
Aa+ button in the style panel.
If coordinates are taken from the ligand, as above, then stabilization calculations are not possible. But coordinates at other locations, such as crystallographic waters, can be used to rapidly produce stabilization results.
> szmap -stbl -at_coords 1dds_waters.oeb.gz -prefix 1dds_xtal_waters \
-p 1DDS_AB__DU__MTX_A-201.oedu
> szmap -stbl -at_coords 1dds_waters.oeb.gz -prefix 1dds_xtal_waters \
-p 1DDS_AB__DU__MTX_A-201_target.oeb.gz -l 1DDS_AB__DU__MTX_A-201_ligand.oeb.gz
GamePlan runs SZMAP at specific coordinates around the binding site and up-to two single bonds away from the ligand, analyzing the results to provide insight into ways to optimize the ligand.
Probe Orientation Data
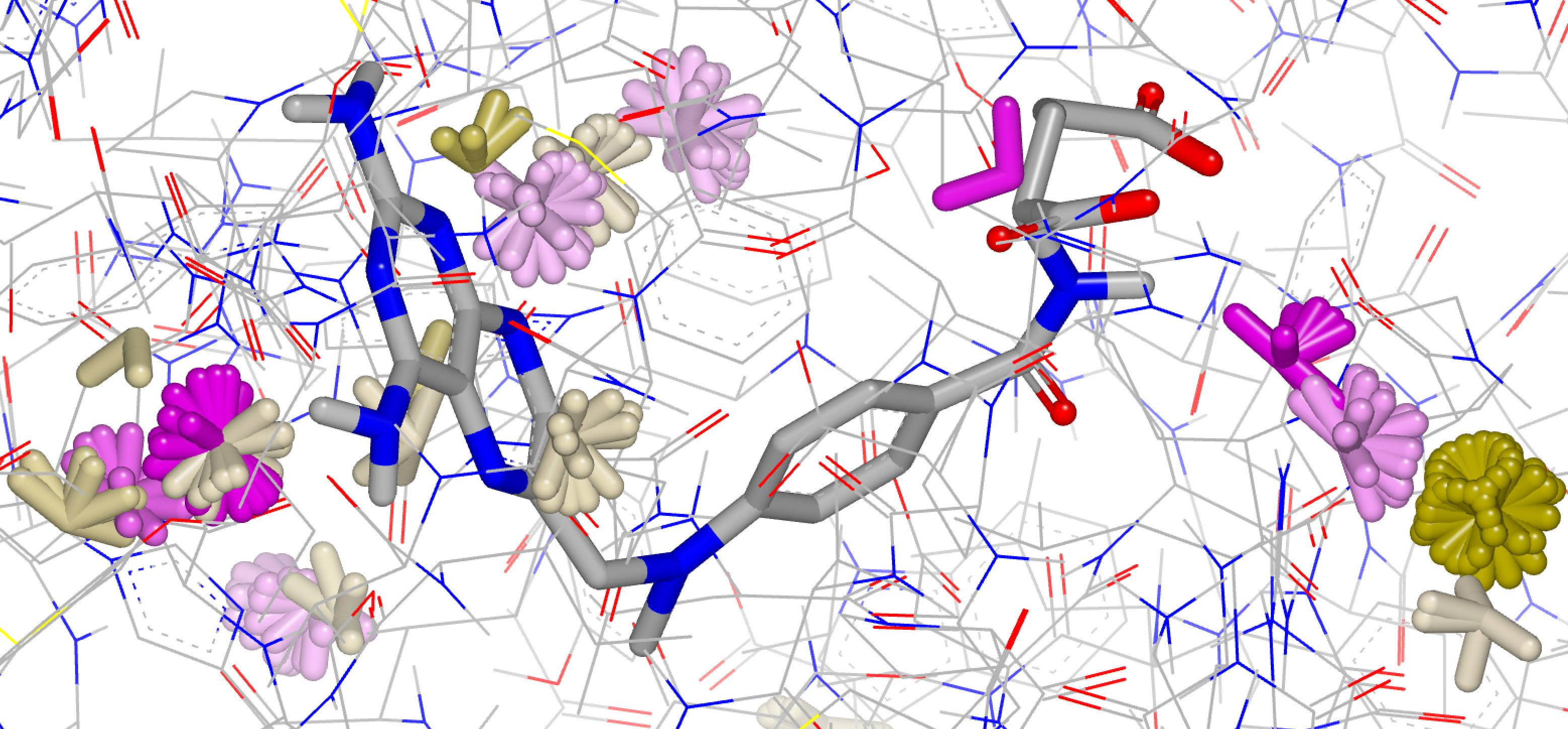
Probe Water Orientations
In both types of SZMAP results, grids and “at coords”, information is stored about the energies and probabilities of the probe for each point. This information can be processed by the Water Orientation VIDA Extension to identify regions of local energy minima and maxima and to display probe order or disorder. The probe geometry at dominant sites in the region where water is displaced can be compared to that of the ligand. High affinity ligands mimic both the difference free energy pattern and the specific geometry of the displaced water. This more “molecular” display is often easier to understand than continuous grids of data and it contains structural information that complements the numerical energy values.