POSIT tutorial
Given receptors, using POSIT is very straightforward. There are two basic ways to input molecules to POSIT.
-in- converts input to 3D conformers ( if 3D structures are input, these initial structures are retained )
-dbase- takes the input conformations as is (these are normally generated with OMEGA.)
For usage of -dbase see POSIT MPI Tutorial.
Given a set of input smiles strings:
> posit -receptor renin/receptors/*.oedu renin/merged/*.oedu -in renin/all.smi
Note
On Microsoft Windows systems, you need to expand the wildcard:
> posit -receptor renin\receptors\2IL2_rec.oedu renin\receptors\2IKU_rec.oedu \ renin\receptors\2IKO_rec.oedu reninmerged\combine_receptors_2IKO_rec_merged_2IKU_rec.oedu \ renin\merged\combine_receptors_2IKU_rec_merged_2IKO_rec.oedu -in renin\all.smi
The following files are output:
posit_docked.oedu- contains all successful poses
posit_score.txt- contains the scores of all successful poses
posit_report.txt- contains the report of the run
posit_status.txt- a periodic status file generated during a run
posit_settings.param- parameters used in the run
The following files are output only if non-empty:
posit_clashed.oeb.gz- contains all poses with good enough probability but clash
posit_undocked.oeb.gz- contains all unsuccessful poses
posit_rejected.txt- list of rejected structures and status of rejection
There is more than one reason a pose may be unsuccessful. The
most common is that the probability of the predicted binding mode is
too low. To control this behavior use the posit -outputall flag, which
writes all output to the file specified by -out in order
of input molecule.
POSIT by default writes docked structure results to .oedu file which can be viewed using VIDA 5. If an
.oeb or .oeb.gz file is desired use the hybrid -docked_molecule_file flag.
To specify the -prefix option to add a prefix to all files output by POSIT or use the -docked_molecule_file option to output a pose file with particular name.
When POSIT is finished, it prints the final status and indicates what new data was added to the results that are output.
The posit_score.txt file contains the scores and ranking of docked structures.
The posit_rejected.txt file can be used to identify the status of rejected molecules,
for instance “All conformers clashed with protein” indicates that while the probability
was good, the protein could not accept the desired pose.
Note
While POSIT can take most molecule formats as input, with large datasets it is fastest to use a pre-generated database of OMEGA [Hawkins-2010] generated conformers. It is recommended, above two rotatable bonds, to generate 100 conformers per rotatable bond when running OMEGA:
> omega2 -in renin/all.smi -out renin/all.oeb.gz -rangeIncrement 1 \ -maxConfRange 200,200,300,400,500,600,700,800,900,1000,1100,1200,1300,1400,1500,1600 > posit -receptor renin/receptors/*.oedu -dbase renin/all.oeb.gz \ -prefix renin
See the posit usage section for more details.
POSIT MPI tutorial
Running POSIT on multiple cores is a simple matter
of adding the -mpi_np argument and specifying the number
of cores desired. When POSIT is run on
a small job as shown above (with 11 molecules and 6) receptors,
using a large number of cores is overkill.
> posit -mpi_np 3 -receptor renin/receptors/*.oedu -dbase renin/all.oeb.gz \ -prefix renin
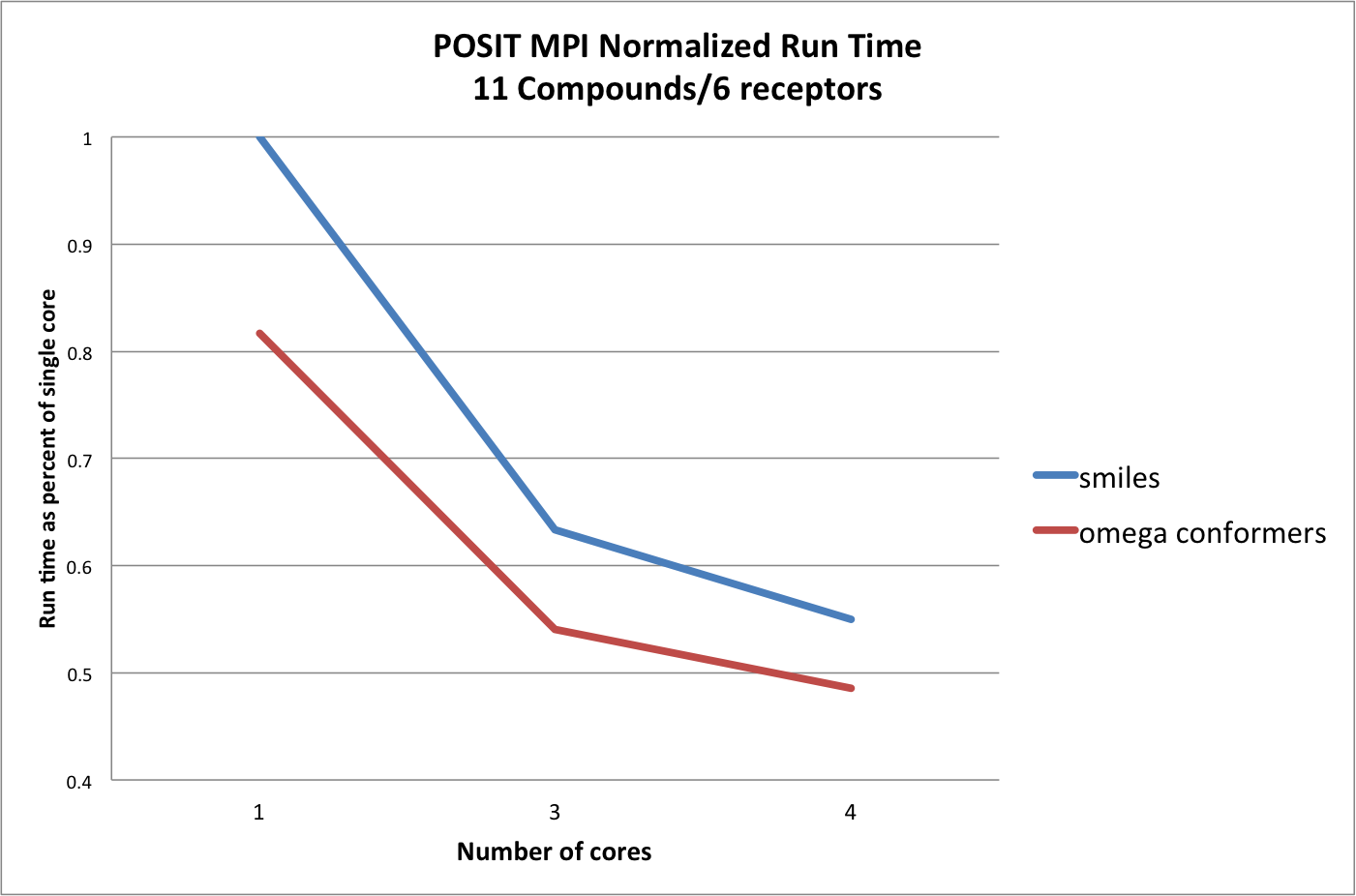
POSIT performance varying the number of cores against a small lead-optimization example.
As seen in figure Posit Performance, running with 3 cores gives a large boost in the run-time and adding another is only marginally faster. Note that running under two cores is not recommended as one core is always the master so, in effect, this is the slowest way to run POSIT.
Also note that using OMEGA conformations as input is the fastest way to run POSIT.