Filter Files
There are two parameter files a user can provide if they would like
to override or augment the default parameter sets. The first is
the “filter file”. It provides acceptable limits for all of the physical
properties and functional groups in the default filter. The second is
the “newrule file”. If you have a filter you like, but would like to
augment it with a set of additional rules, these can be added with a
newrule file.
There are four types of statements that can occur in a filter file:
physical property limits
rules
new rules
selections
The statements should occur one-per-line in the filter file.
Note
If the appropriate line is not in the filter file, or the value is
false, the respective measure will not be used in filtering and
its value will not be included in any table-based output.
Physical Property Limits
There are a large number of physical property limits. They occur as
three fields on a line. For example:
MIN_HETEROATOMS 2 "Minimum number of heteroatoms"
The first field is the property keyword, the second field is the value
assigned to that keyword, and the third field is a brief informational
message. There are a fixed number of physical property keywords. No
additional physical property keywords can be added by the user. The
current keywords and brief definitions of each are listed below.
Hint
The values listed below are those found in the default BlockBuster
filter.
Basic Properties
Molecular Weight
Isotopic molecular weight
MIN_MOLWT 130 "Minimum molecular weight"
MAX_MOLWT 781 "Maximum molecular weight"
Heavy Atom Count
Number of non-hydrogen atoms
MIN_NUM_HVY 9 "Minimum number of heavy atoms"
MAX_NUM_HVY 55 "Maximum number of heavy atoms"
Carbon Count
Number of carbons
MIN_CARBONS 3 "Minimum number of carbons"
MAX_CARBONS 41 "Maximum number of carbons"
Hetero-Count
Number of non-carbon and non-hydrogen atoms
MIN_HETEROATOMS 1 "Minimum number of heteroatoms"
MAX_HETEROATOMS 14 "Maximum number of heteroatoms"
Hetero-Atom to Carbon Ratio
Hetero-count/carbon-count
MIN_Het_C_Ratio 0.04 "Minimum heteroatom to carbon ratio"
MAX_Het_C_Ratio 4.0 "Maximum heteroatom to carbon ratio"
Chiral Count
Number of chiral atoms
MIN_CHIRAL_CENTERS 0 "Minimum chiral centers"
MAX_CHIRAL_CENTERS 21 "Maximum chiral centers"
Hydrogen-bond Acceptors
Number of atoms which match any of the following:
degree 2, aromatic, non-positive nitrogens
electron rich or negative, valence less than 4, non-aromatic nitrogens
negatively charged or not electron withdrawn and neutral oxygens
degree 1, double bonded, electron rich sulfur
MIN_HBOND_ACCEPTORS 0 "Minimum number of hydrogen-bond acceptors"
MAX_HBOND_ACCEPTORS 13 "Maximum number of hydrogen-bond acceptors"
Hydrogen-bond Donors
Number of hydrogen atoms on nitrogen, oxygen, or sulfur atoms
MIN_HBOND_DONORS 0 "Minimum number of hydrogen-bond donors"
MAX_HBOND_DONORS 9 "Maximum number of hydrogen-bond donors"
Lipinski Acceptors
Number of nitrogens or oxygens
MIN_LIPINSKI_ACCEPTORS 1 "Minimum number of oxygen & nitrogen atoms"
MAX_LIPINSKI_ACCEPTORS 14 "Maximum number of oxygen & nitrogen atoms"
Lipinski Donors
Number of nitrogens and oxygens with at least one hydrogen attached
MIN_LIPINSKI_DONORS 0 "Minimum number O & N atoms with hydrogens"
MAX_LIPINSKI_DONORS 6 "Maximum number O & N atoms with hydrogens"
Halide Fraction
Percent of molecular weight from halides
MIN_HALIDE_FRACTION 0.0 "Minimum Halide Fraction"
MAX_HALIDE_FRACTION 0.66 "Maximum Halide Fraction"
Connected Non-Ring
Considers sets of contiguous (bonded) non-ring atoms
MIN_CON_NON_RING 0 "Minimum number of connected non-ring atoms"
MAX_CON_NON_RING 19 "Maximum number of connected non-ring atoms"
Unbranched Chains
The size of unbranched non-ring chains
MIN_UNBRANCHED 1 "Minimum number of connected unbranched non-ring atoms"
MAX_UNBRANCHED 13 "Maximum number of connected unbranched non-ring atoms"
Total Functional Group Count
Total number of functional groups. Does not count any ring-systems as
functional groups. Degree 1 heteroatoms, particularly those with
double bonds or dative bonds are considered part of ring systems and
do not count as a functional group.
MIN_FCNGRP 0 "Minimum number of functional groups"
MAX_FCNGRP 7 "Maximum number of functional groups"
Note
This is different than the functional group rules.
Ring Systems
Number of ring systems (contiguous systems of ring atoms and bonds)
MIN_RING_SYS 0 "Minimum number of ring systems"
MAX_RING_SYS 5 "Maximum number of ring systems"
Ring Size
Maximum size of any single ring system
MIN_RING_SIZE 0 "Minimum atoms in any ring system"
MAX_RING_SIZE 20 "Maximum atoms in any ring system"
Rotor Count
Number of rotatable bonds. Allows optional adjustment for aliphatic
rings following the method of [Oprea-2000].
MIN_ROT_BONDS 0 "Minimum number of rotatable bonds"
MAX_ROT_BONDS 16 "Maximum number of rotatable bonds"
ADJUST_ROT_FOR_RING true "BOOLEAN for whether to estimate degrees of freedom in rings"
Rigid Count
Number of rigid bonds (non-rotatable bonds)
MIN_RIGID_BONDS 4 "Minimum number of rigid bonds"
MAX_RIGID_BONDS 55 "Maximum number of rigid bonds"
LogP
The logP calculation is a derivative of the published XLOGP algorithm
[Wang-R-1997] but is reparameterized without the dependence on 3D
coordinates or the SYBYL/Mol2 aromaticity model.
XLogP
Calculated LogP
MIN_XLOGP -3.0 "Minimum XLogP"
MAX_XLOGP 6.85 "Maximum XLogP"
Solubility
The solubility predictions are based on using the atom types from the
XLOGP algorithm, [Wang-R-1997] and reparameterizing them based on
available solubility data. Rather than a quantitative cutoff, the
solubility uses categories. The six allowable categories are:
insoluble
poorly
moderately
soluble
very
highly
These categories are keywords used in the filter files as follows.
Solubility
Calculated solubility class
MIN_SOLUBILITY insoluble "Minimum solubility"
Pharmacokinetic Predictors
Several secondary filters that are built upon published combinations
of simpler properties are available.
Note
All of these properties are used for filtering in the default
filters.
Lipinski Violations
Number of allowable Lipinski violations. A single Lipinski violation
is considered acceptable. The published work, [Lipinski-1997], allows
compounds to pass with a single violation but not multiple violations.
MAX_LIPINSKI 3 "Maximum number of Lipinski violations"
See also
The Lipinski theory section in the Molecular Properties
and Predictors chapter.
PSA
Peter Ertl’s, [Ertl-2000], topological polar surface area (phosphorus
and sulfur area is optional).
PSA_USE_SandP false "Count S and P as polar atoms"
MIN_2D_PSA 0.0 "Minimum 2-Dimensional (SMILES) Polar Surface Area"
MAX_2D_PSA 205.0 "Maximum 2-Dimensional (SMILES) Polar Surface Area"
See also
The PSA theory section in the Molecular Properties
and Predictors chapter.
GSK/Veber
Veber’s measure of bioavailability (PSA > 140 or Rotatable bonds >10).
[Veber-2002].
GSK_VEBER false "PSA>140 or >10 rot bonds"
Abbott/Martin
Yvonne Martin’s Abbott Bioavailability Score. This is reported as a
probability that F>10% in rats. [Martin-2005]
MIN_ABS 0.11 "Minimum probability F>10% in rats"
Pharmacopia/Egan
Egan egg measure of bioavailability (LogP >5.88 or PSA >
131.6). [Egan-2000]
PHARMACOPIA false "LogP > 5.88 or PSA > 131.6"
Aggregators
Aggregators are small molecules that can interfere with assay results
by sequestering protein in an aggregation of small molecules in
solution. They appear to have activity in many assays, but in fact are
usually not specific inhibitors of the protein in question. Includes
two measures of whether a molecule is one of the aggregators defined
by Shoichet et. al. [McGovern-2003] [Seidler-2003] The first
measure, AGGREGATORS, is whether the molecule is an exact match to
one of the approximately 400 published aggregators. The second
measure, PRED_AGG, is whether the molecule hits in Shoichet’s QSAR
model for predicting aggregators.
Aggregators
Whether a compound is known or predicted to aggregate in
concentrations common in virtual screening.
AGGREGATORS true "Eliminate known aggregators"
PRED_AGG false "Eliminate predicted aggregators"
Elemental Filters
The elemental filters are applied in this order:
Test for the existence of any of the metals in the ELIMINATE_METALS filter in the molecule.
Remove salts by stripping away all the disconnected components except for the largest.
Test to make sure only atoms specified in ALLOWED_ELEMENTS filter are in the molecule.
The format of the two elemental filter fields is the keyword followed
by a comma delimited list of atomic symbols.
Allowed Elements
Molecules with atoms other than those specified by
ALLOWED_ELEMENTS fail to pass the filter.
ALLOWED_ELEMENTS H,C,N,O,F,P,S,Cl,Br,I
Aromatic Ring Count
Uses the result of the function OEGetAromaticRingCount (see toolkit docs) to filter molecules
based on their number of aromatic rings.
MIN_AROMATIC_RING_COUNT 1 "Minimum aromatic ring count"
MAX_AROMATIC_RING_COUNT 6 "Maximum aromatic ring count"
CSP3 Carbon Fraction
Uses the result of the function OEGetFractionCsp3 (see toolkit docs) to filter molecules based
on their CSP3 carbon fraction.
MIN_FRACTION_CSP3 0.1 "Minimum fraction CSP3 carbons"
MAX_FRACTION_CSP3 0.5 "Maximum fraction CSP3 carbons"
Functional Group Rules
Rules statements set the limits for the maximum number of the
specified type of functional group that may be allowed in the
molecule.
The first field of a rule statement is the word RULE in all capital
letters. The second field is a number indicating the maximum number of
the group allowed in a molecule. The third field is the functional
group keyword. Functional-group keywords are case sensitive.
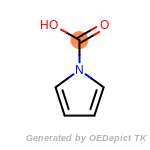
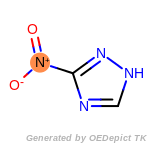
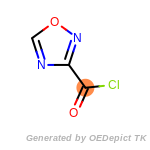
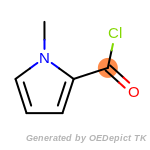
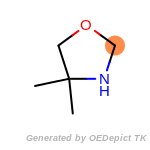
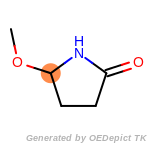
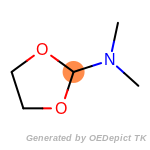
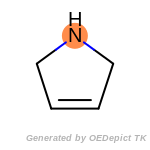
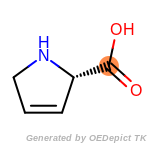
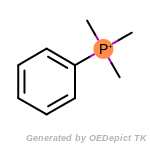
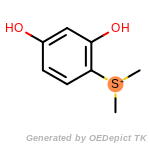
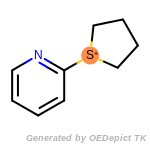
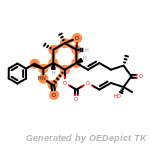
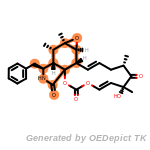
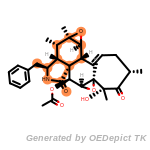
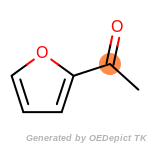
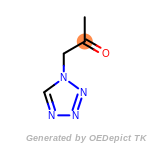
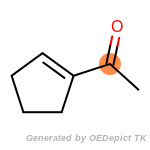
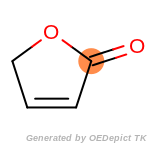
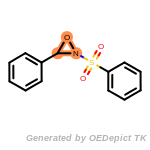
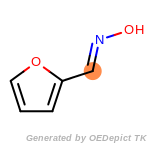
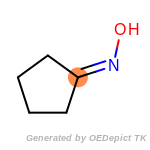
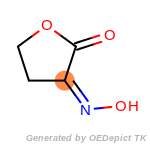
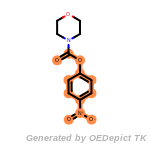
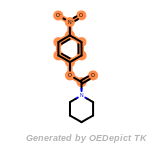
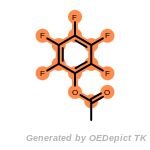
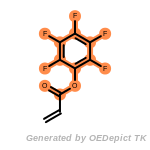
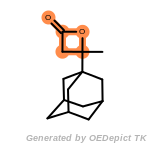
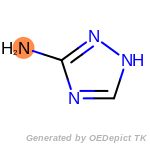
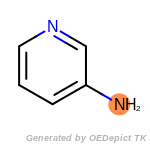
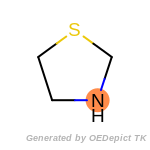
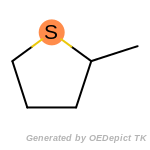
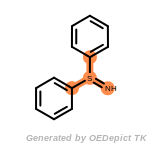
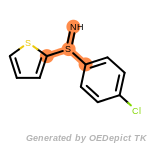
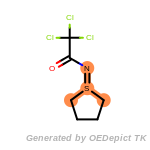
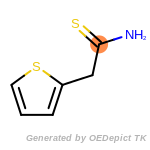
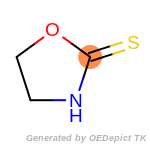
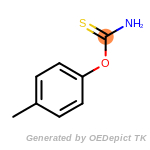
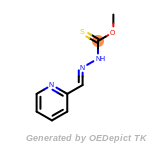
The following is a list of functional groups which filter recognizes
by default. Three example matches are provided with the atoms that
correspond to each other highlighted.
Note
Due to the highly complex nature of the patterns, in particular
recursive SMARTS, it is not possible to fully highlight every atom
that was included as part of the match.
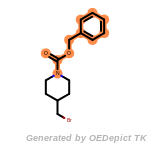
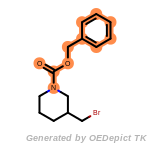
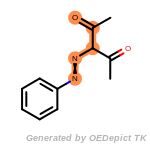
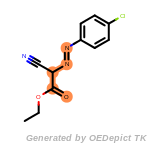
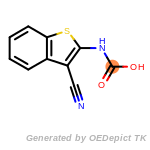
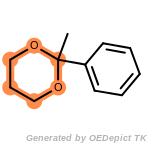
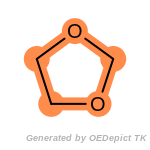
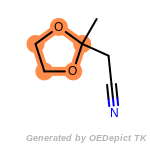
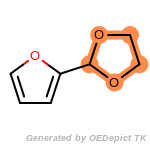
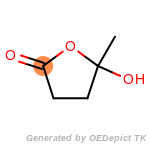
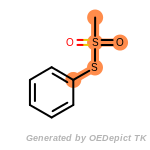
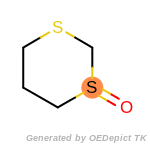
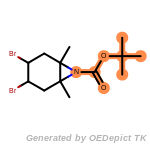
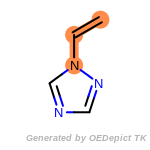
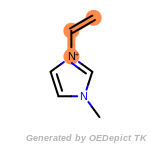
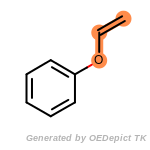
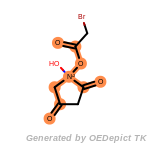
beta_carbonyl_quat_nitrogen
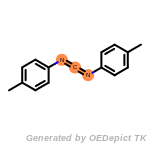
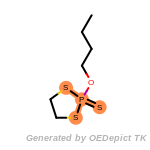
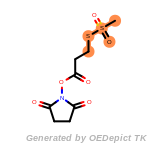
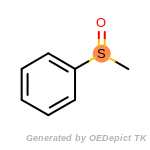
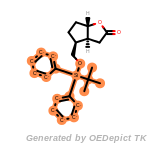
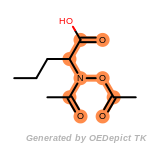
cycloheximide_derivatives
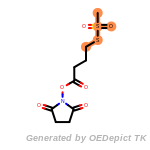
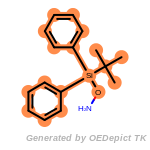
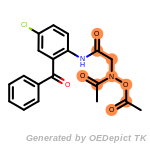
fluorenylmethoxycarbonyl_Fmoc
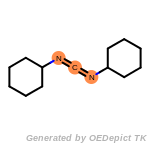
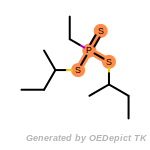
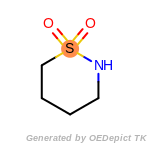
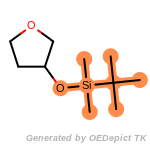
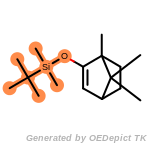
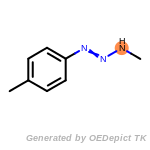
t_butyldimethylsilyl_TBDMS
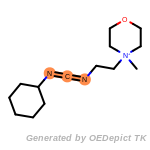
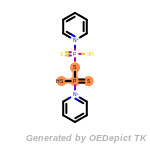
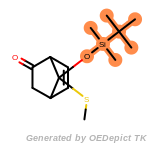
t_butyldiphenylsilyl_TBDPS
New Rules
New rules specify additional functional groups or substructures that
may be used. They must specify a substructure definition in the form
of a SMARTS in addition to the substructure name and maximum
limit. For example:
NEWRULE norbornane 1 C1CC2CCC1C2
The first field is the NEWRULE keyword. The second field defines
the name associated with the substructure (primarily for logging
purposes). The third field indicates the maximum number of the
substructure that can be allowed. The fourth field is the SMARTS
string for the substructure, norbornane in this case. This example rule
would indicate that molecules with a single norbornane substructure
would be allowable, but that those with 2 or more norbornanes would be
eliminated.
New rules that have a name that is identical with one of the original
rules take precedence over the original rule.
Selection Statements
The select statement allows a filter file to specify the required
number of substructures in order to be able to pass the filter. These
statements are similar to new rules except that they list a required
range for passing the filter rather than the range for failing to pass
the filter. For example:
SELECT amine 1 1 [N;!$(*-*[!#6;!#1]);!$(*-a);!$(*=,#*)]
The first field is the SELECT keyword. The second field indicates
the name for the selection (again for logging purposes). The third
field is the minimum number of substructures required to be in the
molecule. The fourth field is the maximum number of substructures
allowed in the molecule. The fifth field is the substructure defined
by a SMARTS pattern. The example requires that molecules contain
exactly one amine. Currently, only a single SELECT statement is
allowed in the filter file. Any complex boolean substructure
statements can be incorporated directly into the SMARTS. If multiple
SELECT statements occur in a filter file, only the final one will
be applied.